
Protezione del gruppo


Un gruppo protettivo ( Inglese gruppo protettore - quindi spesso indicato come abbreviazione comune in regimi formula PG ) è in chimica un sostituente che, durante una più complicato, multi-step sintesi chimica in una molecola è introdotto uno specifico gruppo funzionale per proteggere temporaneamente e quindi una reazione indesiderabile per impedire questo gruppo. Dopo che la reazione desiderata è stata eseguita altrove sulla molecola, il gruppo protettivo viene nuovamente scisso. Per molti gruppi funzionali sono noti diversi possibili gruppi protettivi che differiscono per la loro stabilità e le condizioni per la loro scissione.
Nella sintesi di classi speciali di composti con gruppi funzionali ripetuti - di regola si tratta di biomolecole come peptidi , oligosaccaridi o nucleotidi - sono stati stabiliti set standard di gruppi protettivi. La protezione dei gruppi è diventata oggi uno strumento importante nella sintesi di composti complessi.
I requisiti per un gruppo di protezione sono piuttosto elevati. Ciò include che può essere introdotto specificamente in un gruppo funzionale con una resa molto buona e che deve anche essere in grado di essere nuovamente scisso in condizioni miti. Dovrebbe essere possibile standardizzare le condizioni di reazione per entrambe le fasi. Inoltre, il gruppo protettivo deve essere stabile nel maggior numero possibile di condizioni di reazione. Se possibile, i prodotti di reazione risultanti dovrebbero essere facilmente separabili e, in modo ottimale, anche il reagente del gruppo protettivo è economico. Più ampia è l'esperienza con un gruppo protettivo, migliore è la prevedibilità della reattività del gruppo protettivo.
storia


La storia della tecnologia dei gruppi protettivi è indissolubilmente legata all'uso mirato di diversi composti di partenza per la sintesi di una molecola bersaglio. I primi gruppi protettivi erano generalmente basati sul fatto che il composto di partenza era scelto in modo che un gruppo funzionale reattivo fosse bloccato da un residuo e quindi non reattivo. Così erano z. B. Anisoli scelti al posto di fenoli o esteri invece di gruppi alcolici liberi . È stato solo con la sintesi mirata di composti sempre più complessi emersi dall'inizio del XX secolo che la tecnologia dei gruppi protettivi è diventata davvero importante. A partire dal 1960 circa, si iniziò a investire una notevole spesa per la ricerca nella chimica dei gruppi protettivi. Durante questo periodo, i chimici iniziarono a sintetizzare prodotti naturali sempre più complessi . Particolarmente degni di nota sono i lavori dei vincitori del Premio Nobel Robert B. Woodward , Elias J. Corey e Albert Eschenmoser , che hanno aperto la strada alla sintesi di sostanze naturali complesse.
Oggi esiste un gran numero di gruppi protettivi, che sono riassunti in monografie per quanto riguarda le loro proprietà. Oltre ai gruppi protettivi stabiliti , ci sono molti gruppi protettivi esotici che sono stati sviluppati solo per una sintesi o un'area molto speciale.
Requisiti per un gruppo protezione dati
L'introduzione e la rimozione di gruppi protettivi non rappresentano reazioni produttive in una sequenza di passaggi sintetici; il loro prodotto non si avvicina affatto al prodotto finale desiderato della sintesi. Per questo motivo, vengono spesso posti requisiti elevati sulla protezione delle reazioni di gruppo in termini di prezzo, resa e impegno di sviluppo per la reazione.
Le seguenti caratteristiche sono emerse come requisiti di base per un buon gruppo di protezione:
- Il reagente deve essere disponibile in commercio, poco costoso o facile da produrre
- Il gruppo protettivo deve essere semplice, specifico e in grado di essere introdotto con rese elevate
- Deve essere stabile al maggior numero possibile di condizioni di reazione e metodi di elaborazione e purificazione
- Deve essere specifico, altamente selettivo e in grado di essere suddiviso in rese elevate. Dovrebbe essere possibile standardizzare le condizioni.
- Non deve formare un nuovo stereocentro o un centro diastereotopico
- Dovrebbe essere facilmente riconoscibile negli spettri NMR e interferire il meno possibile attraverso la sovrapposizione del segnale
L'elevata selettività del clivaggio rappresenta un aspetto molto importante, perché spesso i diversi gruppi funzionali devono essere protetti e deprotetti indipendentemente l'uno dall'altro. Idealmente, solo uno dei tanti gruppi protettivi è influenzato dal processo di scissione. Il comportamento dei gruppi protettivi nella pratica non può essere sempre previsto correttamente sulla base della letteratura, soprattutto se in una molecola vengono utilizzati più gruppi protettivi diversi. Pertanto, in alcuni casi, nonostante il bagaglio di esperienza, deve ancora essere svolto un lavoro di sviluppo considerevole durante una sintesi sia per l'introduzione che per il décolleté.
Ortogonalità dei gruppi protettivi

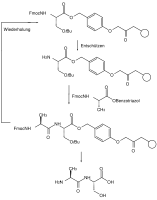
L'ortogonalità dei gruppi protettivi significa che quando vengono utilizzati più gruppi protettivi di diverso tipo, ciascun gruppo protettivo può essere suddiviso individualmente e in qualsiasi ordine sulla base dei vari reagenti di scissione senza attaccare uno degli altri gruppi protettivi. Nell'esempio dell'amminoacido protetto tirosina mostrato , l' estere benzilico può essere suddiviso idrogenoliticamente , il gruppo fluorenilmetilenossi (Fmoc) da basi (ad esempio piperidina ) e l' etere fenolico ter-butilico con acidi (ad esempio acido trifluoroacetico ).
Un esempio diffuso di questa applicazione è la sintesi del peptide Fmoc, che ha acquisito grande importanza sia in soluzione che in fase solida. I gruppi protettivi nella sintesi in fase solida devono essere standardizzati per quanto riguarda le condizioni di reazione come tempo di reazione, temperatura e reagenti in modo che possano essere effettuati da una macchina e si possano ottenere rese ben superiori al 99%, altrimenti la separazione della miscela risultante di prodotti di reazione è praticamente impossibile.

Rappresentazione schematica di una sintesi peptidica in fase solida con gruppi protettivi ortogonali X e Y
Fmoc sintesi peptidica in fase solida con gruppi protettivi ortogonali



Un'altra importante applicazione dei gruppi protettivi ortogonali è nella chimica dei carboidrati . Poiché i carboidrati hanno gruppi idrossilici con reattività molto simile, la protezione o la deprotezione dei singoli gruppi idrossilici deve essere possibile per una conversione sintetica mirata. La sintesi dei nucleotidi rappresenta un caso simile : da un lato c'è il problema (come nella sintesi peptidica) che le molecole sono vettoriali. D'altra parte, c'è anche il problema della chimica dei carboidrati con il residuo zuccherino del ribosio nella sintesi delle molecole di RNA .
Ma anche nella sintesi di sostanze naturali complesse o principi attivi con molti gruppi funzionali si dipende dall'ortogonalità dei gruppi protettivi.
Responsabilità o scissione di gruppi protettivi
Nel caso dei gruppi protettivi, sono state stabilite varie condizioni di reazione che corrispondono al principio di ortogonalità, in base al quale i gruppi protettivi vengono separati. Si può approssimativamente distinguere tra le seguenti condizioni di scissione:
- Gruppi protettivi acido-labili
- Gruppi protettivi labili alla base
- Gruppi protettivi fluoro-labili
- Gruppi protettivi enzimatici labili
- Riduzione: gruppi protettivi labili
- Ossidazione - gruppi protettivi labili
- Gruppi protettivi che sono divisi da sali di metalli pesanti o dai loro complessi
- Gruppi protettivi fotolabili
- Gruppi di protezione a due stadi
I gruppi protettivi acido-labili possono essere separati tramite l'azione degli acidi. La forza trainante qui è spesso la formazione di un carbocatione relativamente stabile o un equilibrio catalizzato da acido che si trova sul lato del gruppo funzionale libero. Esempi di gruppi protettivi acido-labili sono gli esteri terz-butilici , eteri e carbammati, che formano cationi stabili, e gli acetali , in cui, in presenza di acqua, l'equilibrio acido-catalizzato è a fianco delle corrispondenti aldeidi o chetoni.


Nel caso dei gruppi protettivi basolabili, si può fare una distinzione meccanicistica tra l' idrolisi basica e la β-eliminazione indotta dalla base . Gli esteri degli acidi carbossilici (ad eccezione degli esteri terz-butilici ) vengono attaccati nucleofili dagli ioni idrossido e quindi si scindono idroliticamente. Le ammidi, d'altra parte, sono raramente divise in questo modo perché richiedono condizioni molto difficili. Il gruppo ftaloile è un'eccezione , poiché viene scisso con idrazina in condizioni molto miti. Durante la β-eliminazione si verifica una cascata di reazioni: in primo luogo, un protone viene scisso dalla base e si forma un carbanione. Il gruppo protettivo viene quindi tagliato da un adatto gruppo in uscita per formare un composto vinilico . In quest'ultimo caso, il gruppo Fmoc è uno dei più importanti.

Gli ioni fluoruro formano un legame molto stabile con il silicio . Pertanto, i gruppi protettivi del silicio sono quasi senza eccezione tagliati dagli ioni fluoruro. A seconda del tipo di controione o del reagente di scissione, tuttavia, vari gruppi protettivi di silicio possono anche essere tagliati selettivamente in funzione dell'impedimento sterico dell'atomo di silicio. Il vantaggio dei gruppi protettivi fluoro-labili è che nessun altro gruppo protettivo viene attaccato nelle condizioni di scissione.
Gli esteri possono spesso essere scissi da enzimi come le lipasi . Poiché gli enzimi lavorano a un pH compreso tra 5 e 9 ea temperature moderate di circa 30–40 ° C e sono anche molto selettivi per quanto riguarda l'acido carbossilico, questo metodo è un metodo usato raramente, ma molto attraente per scindere i gruppi protettivi.

I gruppi benzilici possono essere scissi in modo riduttivo mediante idrogenazione catalitica . I gruppi benzilici sono usati come eteri, esteri, uretani, carbonati o acetali e sono usati per proteggere alcoli, acidi carbossilici, ammine e dioli.
Sono in uso solo pochi gruppi protettivi che possono essere rimossi mediante ossidazione. Di regola, è metossibenzil etere. Possono essere scissi con cerio (IV) nitrato di ammonio (CAN) o diclorodicanobenzochinone (DDQ) tramite una chinometina .


Il doppio legame di un radicale allilico può essere isomerizzato al composto vinilico da elementi del gruppo del platino (come palladio , iridio o platino ) . Gli enol eteri così ottenuti nel caso di alcoli protetti o enamine nel caso di ammine protette possono essere facilmente idrolizzati in condizioni acide.
I gruppi protettivi fotolabili contengono un cromoforo che può essere attivato per irraggiamento con una lunghezza d'onda adeguata e quindi scisso. Il gruppo o- nitrobenzile (ONB) è elencato qui come esempio .

I gruppi protettivi a due stadi rappresentano una forma speciale di gruppi protettivi , caratterizzati da elevata stabilità, poiché il gruppo protettivo deve essere prima convertito in un gruppo scindibile mediante trasformazione chimica. Tuttavia, questo tipo di gruppo protettivo è usato raramente, poiché qui è necessaria una fase di attivazione aggiuntiva, che estende la sintesi di una fase ulteriore.
Gruppi funzionali
Ammine
Di gran lunga la più grande varietà di gruppi protettivi è disponibile per la funzione amminica . Ciò è dovuto da un lato al fatto che le ammine sono di particolare importanza nella sintesi dei peptidi , ma anche alle loro proprietà: da un lato, sono nucleofili abbastanza potenti , ma anche basi relativamente forti . Queste proprietà hanno portato allo sviluppo di nuovi gruppi protettivi per le ammine.
Molti gruppi protettivi per le ammine sono basati sui carbammati . Questi possono essere facilmente introdotti sotto forma di cloruri di acido carbossilico. Ottengono la loro forza motrice nella scissione dalla formazione della molecola di anidride carbonica molto stabile . Sono state sviluppate varie opzioni di clivaggio sulla base di diversi residui sul carbammato. I carbammati più comunemente usati sono i composti terz- butilossicarbonile, benzilossicarbonile, fluorenilmetilenossicarbonile e allilossicarbonile.
| riposo | formula | Cognome | abbreviazione | scollatura |
|---|---|---|---|---|
| terz- butile |  |
tert -Butyloxycarbonyl | Boc | arrabbiato; Acido trifluoroacetico (TFA) puro o come soluzione in diclorometano , acido cloridrico 3 M in acetato di etile o acido solforico al 10% in diossano |
| Benzile |  |
Benzilossicarbonile | Cbz o Z | idrogenolitico; Idrogeno e palladio su carbone attivo , litio o sodio in ammoniaca liquida |
| Fluorenilmetilene |  |
Fluorenilmetilene ossicarbonile | Fmoc | di base; 20-50% di piperidina in dimetilformammide (DMF) o N-metil-2-pirrolidone , 50% di morfolina in DMF per glicopeptidi sensibili |
| Allyl |  |
Allilossicarbonile | Alloc | scissione catalizzata dal metallo di transizione; Metalli come complessi di palladio (0) o nichel (0) |
Oltre ai carbammati, un certo numero di altri derivati N- acilici sono importanti come gruppi protettivi, ma non sono neanche lontanamente così diffusi. Questi includono, ad esempio, le ftalimmidi , che sono accessibili facendo reagire le ammine primarie con l'anidride ftalica o costruendo il gruppo amminico tramite una sintesi di Gabriel . Le ftalimmidi vengono normalmente scisse dall'idrazina idrato o dal borato di sodio . Le trifluoroacetammidi sono estremamente facili da saponificare nella base , motivo per cui le acetammidi ottenute dalla reazione con l'anidride trifluoroacetica vengono occasionalmente utilizzate come gruppo protettivo per le ammine.
Nel caso di indoli , pirrolo e imidazoli , cioè composti eterociclici, si utilizzano come gruppi protettivi i derivati N- solfonilici. Nel caso delle ammine normali , questo gruppo protettivo è spesso troppo stabile. La rappresentazione qui avviene per solfonazione con fenilsolfonil cloruro e eterociclo deprotonato. La scissione avviene per idrolisi di base. I derivati acilici delle ammine primarie e secondarie sono relativamente facilmente accessibili facendo reagire le ammine con un cloruro di acido aril solfonico, ma possono essere ottenuti solo con difficoltà, per. B. essere suddiviso in condizioni di riduzione della betulla ( sodio in ammoniaca liquida ) o per reazione con naftalide di sodio .
Tra i derivati N -alchilici, i rappresentabili per alchilazione o alchilazione riduttiva hanno derivati N- benzilici di una certa importanza. Come per il gruppo Cbz, la scissione avviene in modo riduttivo e normalmente per idrogenazione catalitica o per riduzione di Betulla. In questo caso, le N- alchilammine hanno lo svantaggio decisivo rispetto ai carbammati o alle ammidi di trattenere l'azoto basico.
Alcoli
Il classico gruppo protettivo per gli alcoli sono gli esteri degli acidi carbossilici . Gli esteri dei precursori sono spesso disponibili in commercio o possono essere facilmente ottenuti facendo reagire gli alcoli con i cloruri acidi o le anidridi mediante una reazione di Schotten-Baumann o mediante transesterificazione . Gli esteri vengono solitamente scissi mediante reazione con nucleofili come idrossidi alcalini, alcolati alcalini o composti organici di litio o magnesio ; in alternativa anche in modo riduttivo per reazione con idruri complessi come litio alluminio idruro . La reattività dell'estere verso l'attacco nucleofilo diminuisce con l'impedimento sterico dell'acido carbossilico nel seguente ordine:
- Cloroacetile> acetile> benzoile> pivaloile
La reattività degli alcoli diminuisce anche con l'aumento dell'impedimento sterico degli alcoli:


Gli esteri più importanti che vengono utilizzati come gruppi protettivi sono gli esteri dell'acido acetico , gli esteri dell'acido benzoico e gli esteri dell'acido pivalico , poiché questi possono essere separati tra loro in modo differenziato a seconda delle reattività date.
I gruppi protettivi più importanti di alcoli e fenoli includono gli eteri di silile trisostituiti molto ben studiati e documentati . Il silicio porta sia gruppi alchilici che arilici come radicali organici. Questo tipo di gruppo protettivo ha il vantaggio di poter essere moderato molto facilmente per quanto riguarda l'introduzione e soprattutto per quanto riguarda il décolleté. Questi eteri vengono prodotti in una sintesi di etere Williamson dal clorosilano e uno ione alcolato o utilizzando reagenti di attivazione come l' imidazolo .
Per scopi puramente analitici, ad es. B. per rendere volatile un carboidrato ed essere in grado di rilevarlo con l'aiuto di GC-MS , esistono kit di reazione disponibili in commercio. Gli eteri di silile sono generalmente sensibili agli acidi e agli ioni fluoruro. Quest'ultimo è principalmente utilizzato per dividerli. Tuttavia, i prezzi commerciali dei clorosilani variano notevolmente a seconda della sostituzione. Il clorosilano più economico qui è il clorotrimetilsilano (TMS-Cl), che è un sottoprodotto della produzione di silicone secondo Rochow e Müller . Un'altra fonte comune del gruppo trimetilsilile è l' esametildisilazano (HMDS). Tuttavia, gli eteri trimetilsililici sono anche estremamente sensibili all'idrolisi acida (ad esempio il gel di silice è sufficiente come donatore di protoni) e sono quindi usati raramente come gruppo protettivo oggi.
| Cognome | formula | abbreviazione | scollatura |
|---|---|---|---|
| Trimetilsilile |  |
TMS | Fluoruro di potassio , acido acetico o carbonato di potassio in metanolo |
| Trietilsilile |  |
TES | 10-100 volte più stabile di un gruppo TMS; Acido trifluoroacetico in acqua / tetraidrofurano , acido acetico in acqua / tetraidrofurano, acido fluoridrico , piridinio idrofluoruro in piridina |
| terz- butildimetilsilile |  |
TBS, TBDMS | Acido acetico in tetraidrofurano / acqua, piridinio tosilato in metanolo, acido trifluoroacetico in acqua, acido fluoridrico in acetonitrile , piridinio idrofluoruro in tetraidrofurano, tetrabutilammonio fluoruro in THF |
| Triisopropilsilile |  |
SUGGERIMENTI | Nelle stesse condizioni di TBS ma tempi di reazione più lunghi; Fluoruro di tetrabutilammonio in tetraidrofurano, acido fluoridrico in acetonitrile, fluoruro di piridinio in tetraidrofurano. |
| terz -Butyldiphenylsilyl |  |
TBDPS | Nelle stesse condizioni di TBS ma tempi di reazione più lunghi (100-250 volte più lenti di TBS e 5-10 volte più lenti di TIPS); Fluoruro di tetrabutilammonio in tetraidrofurano, acido fluoridrico in acetonitrile, fluoruro di piridinio in tetraidrofurano |
Un'altra classe di gruppi protettivi per gli alcoli sono gli eteri alchilici. Anche qui ci sono possibilità varie e ortogonali per dividere gli eteri. Gli eteri metossi alifatici sono difficili da scindere e in condizioni drastiche, quindi sono generalmente usati solo con i fenoli.
| Cognome | formula | abbreviazione | scollatura |
|---|---|---|---|
| metile | Me | Solitamente utilizzato solo per fenoli; Iodotrimetilsilano in cloroformio , diclorometano o acetonitrile, tribromuro di boro o tricloruro di boro in diclorometano, acidi di Lewis (cloruro di alluminio, trifluoruro di boro in presenza di tioli) | |
| Benzile |  |
Bn | riduttivo; Idrogenazione catalitica (palladio su carbone attivo, nichel Raney o rodio su ossido di alluminio come catalizzatore) |
| p- metossibenzile | PMB, MPM | ossidativo; DDQ (diclorodicyanoquinone) in diclorometano, cloruro di ammonio cerico in acqua | |
| 3,4-dimetossbenzile |  |
DMB, DMPM | come il PMB ossidativo; DDQ (diclorodicyanoquinone) in diclorometano, cloruro di ammonio cerico in acqua |
| Trifenilmetile (tritile) |  |
Tr | arrabbiato; Acido formico in etere o acqua, 80% di acido acetico, 1 M di acido cloridrico |
| terz- butile |  |
arrabbiato; acido trifluoroacetico anidro, acido bromidrico / acido acetico, acido cloridrico 4N | |
| Allyl | Potassio terz- butanolato, palladio su carbone attivo, DABCO in metanolo, vari complessi di elementi di platino - quindi lavorazione acida. | ||
| Allilossicarbonile | Alloc | Come allile; Potassio terz- butanolato, palladio su carbone attivo, DABCO in metanolo, vari complessi di elementi di platino - quindi lavorazione acida | |
| Metossimetile | MAMMA | Arrabbiato; Acido cloridrico 6 M in tetraidrofurano / acqua | |
| Methylthiomethyl | MTM | Cloruro di mercurio (II) / carbonato di calcio in acetonitrile / acqua, nitrato d'argento in tetraidrofurano / acqua | |
| (2-metossietossi) metile | MEM | Acido bromidrico acquoso in tetraidrofurano, bromuro di zinco in diclorometano | |
| Benzilossimetile | BOM | Paragonabile alla stabilità di MOM, MEM e SEM; Riduttivo; Sodio in ammoniaca liquida, idrogenazione catalitica (idrossido di palladio su carbone attivo), nichel Raney in etanolo | |
| β- (trimetilsilil) etossimetile |  |
SEM | Più labile di MEM e MOM nei confronti dell'idrolisi acida; Acido cloridrico 0,1 M in metanolo, acido fluoridrico concentrato in acetonitrile, trifluoruro di boro eterato in diclorometano, fluoruro di tetrabutilammonio in HMPT ( esametilfosforico triamide dell'acido) o tetraidrofurano |
| Tetraidropiranile |  |
THP | Acido acetico in tetraidrofurano / acqua, acido p- toluensolfonico in metanolo |
1,2-dioli
Gli 1,2-dioli ( glicoli ) rappresentano una classe speciale di alcoli nella protezione della chimica di gruppo . B. nel caso degli zuccheri , sfruttare il fatto che entrambi i gruppi idrossilici sono protetti come acetali dipendenti l'uno dall'altro . Comunemente usati qui sono gli acetali benzilidene , isopropilidene e cicloesilidene o ciclopentilidene .

Gli acetali vengono generalmente prodotti spostando l'equilibrio di una miscela del glicole con il componente carbonilico rimuovendo l'acqua di reazione o mediante transacetalizzazione con un semplice acetale e rimuovendo l'alcol formato dalla miscela di reazione.

Nella chimica dello zucchero in particolare, le diverse posizioni dei gruppi idrossilici l'uno rispetto all'altro vengono utilizzate per proteggerli selettivamente in una certa dipendenza stereochimica. Oltre alle altre possibili combinazioni, i due gruppi ossidrilici adiacenti che formano la conformazione più stabile reagiscono preferenzialmente tra loro.

In linea di principio, gli acetali possono essere nuovamente scissi in solventi acidi acquosi. Il gruppo protettivo benzilidene, che può anche essere suddiviso in modo riduttivo, è un caso speciale qui. Questo viene fatto mediante idrogenazione catalitica o dal donatore di idruro diisobutil alluminio idruro (DIBAL). Tuttavia, la scissione di DIBAL deprotige solo un gruppo alcolico, poiché il residuo benzilico rimane sul secondo gruppo idrossile e più stericamente ostacolato rispetto all'etere benzilico.

Gruppi carbonilici
I gruppi carbonilici sono principalmente a rischio di attacchi nucleofili come i reagenti di Grignard o gli ioni idruro. Le aldeidi possono anche essere ossidate ad acidi carbossilici. Ma anche reazioni indesiderate causate da reazioni catalizzate da acidi e basi del gruppo carbonile come tali. B. Le reazioni aldoliche possono essere prevenute da un gruppo protettivo adatto.
I gruppi protettivi più comuni per i gruppi carbonilici sono gli acetali e soprattutto gli acetali ciclici con dioli. Inoltre, vengono utilizzati anche acetali ciclici con 1,2-idrossitioli o ditioglicoli, i cosiddetti acetali O , S o S , S.


Per gli acetali come gruppo protettivo per i composti carbonilici, lo stesso vale in linea di principio per gli acetali come gruppo protettivo per 1,2-dioli. Sia la produzione che la scollatura sono naturalmente identiche. Tuttavia, con gli acetali come gruppo protettivo, il processo di transacetalizzazione gioca un ruolo subordinato e di solito sono prodotti dai glicoli scindendo l'acqua. Varianti più moderne utilizzano anche glicoli in cui gli atomi di idrossi idrogeno sono stati sostituiti da un gruppo trimetilsilile. Normalmente, come dioli per gli acetali vengono utilizzati glicoli semplici come il glicole etilenico o l' 1,3-propandiolo .
Gli acetali possono essere scissi in condizioni acquose acide. Gli acidi minerali sono usati come acidi. Il cosolvente è spesso acetone , che viene utilizzato come solubilizzante. Come metodo di eliminazione non acido, è possibile utilizzare un complesso cloruro- acetonitrile di palladio (II) in acetone o cloruro di ferro (III) su gel di silice assorbito in cloroformio.
Gli acetali ciclici sono molto più stabili all'idrolisi acida rispetto agli acetali aciclici. Pertanto, gli acetali aciclici vengono utilizzati quasi esclusivamente se è necessaria una scissione molto lieve o se due diversi gruppi carbonilici protetti devono essere differenziati per quanto riguarda il loro rilascio.
Tuttavia, oltre alla loro unica funzione di gruppo protettivo, gli acetali sono anche usati come reagenti ausiliari chirali . Quindi acetali di glicoli chirali come. B. i derivati dell'acido tartarico possono essere aperti in modo asimmetrico con elevata selettività. Ciò consente la creazione di nuovi centri di chiralità.

Oltre agli acetali O , O- , anche gli acetali S , O - e S , S hanno un'importanza, sebbene minore, come gruppo protettivo del carbonile. I tioli , che devono essere utilizzati per la produzione di questi acetali, hanno un odore molto sgradevole e sono velenosi, il che ne limita molto l'uso. I tioacetali e gli acetali S , O misti sono, rispetto agli acetali O , O puri, molto più stabili all'idrolisi acida. Ciò consente la scissione selettiva di questi gruppi carbonilici in presenza di gruppi carbonilici protetti dallo zolfo .
Gli acetali S , S sono normalmente prodotti in modo analogo agli acetali O , O per catalisi acida dai ditioli e dal componente carbonilico. A causa della grande stabilità dei tioacetali, l'equilibrio è dalla parte degli acetali. A differenza degli acetali O , O , non è necessario rimuovere l'acqua di reazione per spostare l'equilibrio.
Gli acetali S , O vengono idrolizzati 10.000 volte più velocemente dei corrispondenti acetali S , S. Sono prodotti in analogia a questi dai tioalcoli. La loro scissione avviene anche in condizioni comparabili e principalmente attraverso composti di mercurio (II) in acetonitrile acquoso.
Per le aldeidi è stata descritta una protezione temporanea del gruppo carbonile in presenza di chetoni come anione emiaminale . Ciò sfrutta il fatto che le aldeidi hanno un'attività carbonilica molto più elevata rispetto ai chetoni e che molte reazioni di addizione sono reversibili.

Gruppi carbossi
I gruppi protettivi più importanti per i gruppi carbossilici sono gli esteri di vari alcoli. Vengono utilizzati anche orto -esteri e ossazoline , ma di minore importanza. Esistono fondamentalmente diversi metodi per la produzione di esteri di acidi carbossilici:
- Esterificazione diretta di acidi carbossilici e componenti alcolici. A causa dell'equilibrio sfavorevole nella reazione tra alcoli e acidi carbossilici, l'equilibrio deve essere raggiunto rimuovendo l'acqua di reazione o lavorando con grandi eccessi di alcol. Per fare questo, però, l'alcol deve essere molto economico. Questa reazione è catalizzata da acido (acido solforico, acido p- toluensolfonico o scambiatori di ioni acidi sono i catalizzatori di esterificazione più comuni).
- La reazione di anidridi acide o cloruri acidi con alcoli in presenza di basi ausiliarie. La piridina , la diisopropiletilammina o la trietilammina sono spesso utilizzate qui come basi ausiliarie . Questa reazione può essere catalizzata con 4- N , N- dimetilamminopiridina , che aumenta la velocità di reazione di un fattore 10 4 rispetto alla piridina pura . Rispetto all'esterificazione diretta, questi metodi vengono eseguiti in condizioni molto blande.
- La reazione dei sali di acidi carbossilici con alogenuri alchilici è un altro metodo per la preparazione di esteri di acidi carbossilici.
- La reazione degli acidi carbossilici con l' isobutene è un metodo delicato per produrre esteri ter-butilici . Qui l'isobutene viene fatto reagire con l'acido carbossilico in presenza di un acido forte come l' acido solforico .
- La reazione degli acidi carbossilici con i diazoalcani è un metodo molto delicato e quantitativo per produrre esteri. Tuttavia, a causa della scarsa accessibilità dei diazoalcani complessi, viene utilizzato principalmente solo per la produzione di esteri metilici e benzidrilici.
Oltre a questi metodi classici di esterificazione, sono stati sviluppati altri e più moderni metodi per reazioni speciali.
- L'attivazione dell'acido carbossilico con la dicicloesilcarbodiimmide e la reazione dell'O -acilisourea così ottenuta con la componente alcolica in presenza di 4- N , N- dimetilamminopiridina ( esterificazione di Steglich ).
- Attivazione dell'acido carbossilico mediante produzione di un'anidride mista con acido 2,4,6-triclorobenzoico mediante reazione dell'acido carbossilico con cloruro di benzoile in presenza di 4- N , N- dimetilamminopiridina e trietilammina. L'anidride miscelata viene prodotta in situ e fatta reagire immediatamente ulteriormente con la componente alcolica ( esterificazione Yamaguchi ).
- L'attivazione della componente alcolica attraverso la reazione in condizioni Mitsunobu con dietilazodicarbossilato e trifenilfosfina e successiva reazione in situ con l'acido carbossilico ( esterificazione Mitsunobu ).
Vari gruppi possono servire come componente alcolico. Tuttavia, gli esteri metilici, terz - butilici, benzilici e allilici sono molto comuni qui . Inoltre, ci sono anche una serie di gruppi protettivi, che derivano dai gruppi protettivi eterei dei gruppi idrossilici. Tuttavia, le condizioni di scissione specifiche sono spesso molto simili. In linea di principio, qualsiasi estere può essere idrolizzato in presenza di ioni idrossido in una soluzione acquoso-alcolica. Per substrati più sensibili, invece, viene spesso utilizzato idrossido di litio in tetraidrofurano e in presenza di metanolo. Le stesse regole valgono naturalmente per la tendenza all'idrolisi come per gli esteri come gruppi protettivi dell'alcol.
| Cognome | formula | abbreviazione | scollatura | Fabbricazione speciale |
|---|---|---|---|---|
| metile | Me | nucleofilo-alcalino da idrossidi metallici o carbonati in metanolo acquoso o tetraidrofurano, alogenuri di metalli alcalini in solventi aprotici umidi come dimetilsolfossido, N , N- dimetilformammide nel calore, enzimaticamente z. B. da esterasi epatica di maiale | Diazometano in etere dietilico, carbonato di cesio e ioduro di metile in N , N- dimetilformammide, metanolo e trimetilsilil cloruro catalitico | |
| terz- butile | |
tert -Bu | arrabbiato; Acido trifluoroacetico (puro o in diclorometano), acido formico, acido p- toluensolfonico | Isobutene in diossano e acido solforico catalitico |
| Benzile | |
Bn | idrogenolitico; Idrogeno / palladio su carbone attivo | |
| Benzidrile |  |
idrogenolitico; Idrogeno / palladio su carbone attivo (molto facile da dividere) | ||
| Allyl | Allyl | analogo agli eteri; Potassio terz- butanolato, palladio su carbone attivo, DABCO (1,4-diazabiciclo [2.2.2] ottano) in metanolo, vari complessi di elementi platino - quindi lavorazione acida |
Alcheni
Gli alcheni sono e raramente devono essere protetti da un gruppo protettivo. Di regola, sono influenzati solo da attacchi elettrofili , isomerizzazione e, durante l'idrogenazione catalitica, da reazioni collaterali indesiderate. Fondamentalmente, due gruppi protettivi sono noti per gli alcheni:
- Alogenazione temporanea con bromo per formare il composto trans -1,2-dibromoalchilico: l'alchene viene rigenerato ripristinando la conformazione con zinco elementare o con dicloruro di titanocene .
- Protezione attraverso una reazione Diels-Alder : la reazione di un alchene con un diene porta ad un alchene ciclico, che è a rischio di attacco elettrofilo in modo simile all'alchene originale. Il diene utilizzato come gruppo protettivo viene scisso termicamente, poiché una reazione di Diels-Alder è una reazione reversibile o di equilibrio.

Alkynes
Sono noti anche due tipi di gruppi protettivi per gli alchini. Con gli alchini terminali a volte è necessario mascherare l'atomo di idrogeno acido. Ciò avviene normalmente mediante deprotonazione (utilizzando basi forti come metilmagnesio bromuro o butillitio in tetraidrofurano / dimetilsolfossido ) e successiva reazione con clorotrimetilsilano per formare l'alchino terminale protetto da TMS. La scissione avviene idroliticamente - con carbonato di potassio in metanolo - o da ioni fluoruro come, ad esempio, per mezzo di fluoruro di tetrabutilammonio .

Per proteggere il triplo legame stesso, a volte viene utilizzato un complesso del composto alchino con dicobalto ottacarbonile . Il cobalto viene scisso per ossidazione.

Applicazioni
I gruppi protettivi sono utilizzati in un'ampia gamma di chimica organica sintetica. Questo vale sia per le sintesi di laboratorio che per le sintesi su larga scala di principi attivi complessi. Non appena un gruppo funzionale si rivela distruttivo o può essere attaccato in modo indesiderato, viene utilizzata la tecnica del gruppo protettivo. I gruppi protettivi sono usati in quasi tutte le sintesi di una molecola bersaglio complessa. Poiché l'introduzione oltre che la scollatura dei gruppi protettivi comporta sia lo sforzo che una perdita di resa, è auspicabile farne a meno i gruppi protettivi, cosa che però spesso è difficile da ottenere.
Nella sintesi automatizzata di peptidi e nucleotidi, la protezione della chimica dei gruppi è parte integrante del concetto di sintesi. I gruppi protettivi sono anche indispensabili nella chimica dello zucchero a causa dei gruppi idrossilici molto simili nelle molecole di zucchero.
Un importante esempio dell'applicazione industriale della tecnologia dei gruppi protettivi è la sintesi dell'acido ascorbico (vitamina C) secondo Reichstein .

Al fine di prevenire l' ossidazione degli alcoli secondari da parte del permanganato di potassio , essi vengono protetti mediante acetalizzazione con acetone e nuovamente deprotetti dopo l'ossidazione del gruppo idrossile primario ad acido carbossilico.
Un esempio molto spettacolare dalla sintesi di prodotti naturali per l'applicazione di gruppi protettivi è la sintesi totale dell'acido carbossilico della palitossina da parte del gruppo di lavoro di Yoshito Kishi nel 1994. 42 gruppi funzionali (39 gruppi idrossilici, un diolo, un gruppo amminico e un acido carbossilico gruppo) doveva essere protetto. Ciò è stato fatto da otto diversi gruppi protettivi (un estere metilico, cinque gruppi acetato, 20 TBDMS eteri, nove p- metossibenzil eteri, quattro benzoati, un metil emiacetale, un acetale con acetone e un SEM estere).

L'introduzione o la modifica di un gruppo protettivo a volte influenza anche la reattività dell'intera molecola. A titolo di esempio, qui viene mostrato un estratto dalla sintesi di un analogo della mitomicina C di Danishefsky .

Cambiare il gruppo protettivo da un etere metilico a un etere MOM impedisce all'epossido di aprirsi per formare l' aldeide .
Un'importante applicazione della chimica dei gruppi protettivi può essere trovata nella sintesi automatizzata di peptidi e nucleosidi. Nella sintesi peptidica mediante sintetizzatori automatici, vengono utilizzate l'ortogonalità del gruppo Fmoc (scissione di base), il gruppo terz - butile (scissione acida) e vari gruppi protettivi per gruppi funzionali nella catena laterale degli amminoacidi. Nella sintesi nucleotidica automatizzata di sequenze di DNA e RNA dopo la sintesi di fosforamidite , vengono utilizzati fino a quattro diversi gruppi protettivi per blocco di costruzione. Qui anche la chimica redox avviene sugli atomi di fosforo protetti. Il fosforo trivalente utilizzato per la sua maggiore reattività è dotato di un gruppo protettivo cianoetilico sull'ossigeno libero. La fase di accoppiamento è seguita dall'ossidazione al fosfato, trattenendo il gruppo protettivo. I gruppi OH liberi che non hanno reagito nella fase di accoppiamento vengono acetilati in una fase intermedia. Questo gruppo protettivo aggiunto in aggiunta impedisce quindi a questo gruppo OH di accoppiarsi nei cicli successivi.

Di regola, l'introduzione di un gruppo protettivo non è un problema. Le difficoltà risiedono più nella loro stabilità e nella divisione selettiva. I problemi che sorgono nelle strategie di sintesi con gruppi protettivi sono raramente documentati nella letteratura specialistica.
Economia atomica
Le sintesi che utilizzano gruppi protettivi generalmente hanno una bassa economia atomica . A volte la deviazione dall'uso di gruppi protettivi deve essere presa per eliminare reazioni concorrenti indesiderate e per ottenere la selettività desiderata di una sintesi. La protezione delle strategie di gruppo è spesso indispensabile nella sintesi di strutture complesse.
Le sintesi di Hapalindol U vengono confrontate come esempio di una strategia di gruppo protettore rispetto a una sintesi priva di gruppi protettivi . Mentre la sintesi di Hideaki Muratake del 1990 utilizzava tosyl come gruppo protettivo, nella sintesi di Phil S. Baran del 2007 non è stato utilizzato alcun gruppo protettivo. Il numero di passaggi di sintesi è stato notevolmente ridotto.

Hapalindol U Muratake 1990 Ts sintesi del gruppo protettivo (gruppi protettivi in blu .)

Hapalindol U Baran 2007 Sintesi senza gruppi di protezione


letteratura
- Philip J. Kocieński : Protecting Groups , 1a edizione, Georg Thieme Verlag, Stoccarda 1994, ISBN 3-13-135601-4 .
- Peter GM Wuts, Theodora W. Greene: Green's Protective Groups in Organic Synthesis , 4th Ed., John Wiley & Sons Inc., Hoboken, New Jersey, ISBN 0-471-69754-0 .
- Michael Schelhaas, Herbert Waldmann : "Protecting Group Strategies in Organic Synthesis", in: Angewandte Chemie , 1996 , 103 , pp. 2192–2219; doi: 10.1002 / anie.19961081805 .
- Krzysztof Jarowicki, Philip Kocieński: "Protecting groups" , in: J. Chem. Soc., Perkin Trans. 1 , 1998 , pp. 4005-4037; doi: 10.1039 / A803688H .
link internet
- Università di Marburg : gruppi di protezione nella chimica organica sintetica (PDF; 284 kB)
- Organic-Reaction.com: protezione del gruppo
- Organic-Chemistry.org: protezione dei gruppi
Prove individuali
- ↑ a b Kyriacos C. Nicolaou, Erik J. Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods , 1996, ISBN 3-527-29284-5 .
- ↑ a b c Kyriacos C. Nicolaou, Scott A. Snyder: Classics in Total Synthesis II , Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2003, ISBN 3-527-30684-6 .
- ↑ Philip J. Kocienski: Protecting Groups , 1a edizione, Georg Thieme Verlag, Stoccarda 1994, ISBN 3-13-135601-4 .
- ↑ Peter GM Wuts, Theodora W. Greene: Green's Protective Groups in Organic Synthesis , Fourth Ed. John Wiley & Sons Inc., Hoboken, New Jersey, ISBN 0-471-69754-0 .
- ↑ PJ Kocienski: Protecting Groups , pagine 245-250.
- ^ Dietrich Spitzner, Kai Oesterreich: "Reazioni di domino indotte anionicamente - Sintesi di un terpene di tipo Norpatchoulenol", in: European Journal of Organic Chemistry , 2001 , 10 ; Pp. 1883-1886; doi : 10.1002 / 1099-0690 (200105) 2001: 10 <1883 :: AID-EJOC1883> 3.0.CO; 2-M .
- ↑ a b c Weng C. Chan, Peter D. White: Fmoc Solid Phase Peptide Synthesis . Ristampa 2004, Oxford University Press, ISBN 0-19-963724-5 .
- ↑ Weng C. Chan, Peter D. White: Fmoc Solid Phase Peptide Synthesis , pp. 10-12.
- ↑ Kyriacos C. Nicolaou, Eric J. Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods , VCH Verlagsgesellschaft mbH, Weinheim, 1996, pp. 711-729, ISBN 3-527-29284-5 .
- ↑ Michael Schelhaas, Herbert Waldmann: "Strategie di gruppo di protezione nella sintesi organica", in: Angewandte Chemie , 1996 , 103 , pp. 2195-2200; doi: 10.1002 / anie.19961081805 .
- ^ VN Rajasekharan Pillai: "Gruppi protettivi fotoreportabili nella sintesi organica", in: Synthesis , 1980 , pp. 1-26.
- ↑ PJ Kocienski: Protecting Groups , S. 186th
- ↑ Naomi Sakai, Yasufumi Ohfune: "Sintesi totale di galantina I. Ciclizzazione catalizzata da acido dell'acido galantinico", in: J. Am. Chem. Soc. , 1992 , 114 , pagg. 998-1010; doi: 10.1021 / ja00029a031 .
- ↑ Glenn L. Stahl, Roderich Walter, Clarck W. Smith: "Procedura generale per la sintesi di 1,6-diaminoesani mono-N-acilati", in: J. Org. Chem. , 1978 , 43 , p. 2285– 2286; doi: 10.1021 / jo00405a045 .
- ^ RA Houghton, A. Beckman, JM Ostresh: Int. J. Pept. Protein Res. , 1986 , 27 , p. 653.
- ↑ PJ Kocienski: Protecting Groups , S. 195th
- ^ Robert M. Williams, Peter J. Sinclair, Dongguan Zhai, Daimo Chen: "Sintesi asimmetriche pratiche di α-amminoacidi attraverso costruzioni di legami carbonio-carbonio su modelli di glicina elettrofila", in: J. Am. Chem. Soc. , 1988 , 110 , pagg. 1547-1557; doi: 10.1021 / ja00213a031 .
- ↑ Weng C. Chan, Peter D. White: Fmoc Solid Phase Peptide Synthesis , pp. 27-30.
- ^ Campi Gregg B.: metodi per rimuovere il gruppo Fmoc. (PDF; 663 kB) In: Michael W. Pennington, Ben M. Dunn (a cura di): Peptide Synthesis Protocols Volume 35, 1995, ISBN 978-0-89603-273-6 , pp. 17-27.
- ↑ B. Liebe, H. Kunz: sintesi in fase solida di un glicopeptide antigene sialyl-Tn associato al tumore con una sequenza parziale dalla "ripetizione in tandem" della mucina MUC-1 In: Angew. Chem. Vol. 109, 1997, pagg. 629-631.
- ↑ ChemPep Inc.: Fmoc Solid Phase Peptide Synthesis. Estratto il 16 novembre 2013.
- ↑ PJ Kocienski: Protecting Groups , pagine 199-201.
- ↑ John O. Osby, Michael G. Martin, Bruce Ganem: An Exceptionally Mild Deprotection of Phthalimides , in: Tetrahedron Lett. , 1984 , 25 , pagg. 2093-2096; doi: 10.1016 / S0040-4039 (01) 81169-2 .
- ↑ PJ Kocienski: Protecting Groups , pagine 220-227.
- ↑ P. Vouros: "Derivatizzazione chimica nella gascromatografia-spettrometria di massa", in: "Mass Spectrometrie", Degger, New York, 1979, vol. 2, p. 129.
- ↑ PJ Kocienski: Protecting Groups , S. 29th
- ↑ PJ Kocienski: Protecting Groups , S. 31st
- ↑ Tod K Jones, Robert A. Reamer, Richard Desmond, Sander G. Mills: "Chemistry of tricarbonyl hemiketals and application of Evans technology to the total synthesis of the immunosoppressant (-) - FK-506", in: J. Am. Chem. Soc. , 1990 , 112 , pagg. 2998-3017; doi: 10.1021 / ja00164a023 .
- ↑ Dieter Seebach, Hak-Fun Chow, Richard FW Jackson, Marius A. Sutter, Suvit Thaisrivongs, Jürg Zimmermann: "(+) - 11,11′-Di-O-methylelaiophylidene - preparazione da elaiophylin e sintesi totale da (R) -3-idrossibutirrato e (S) -malato ", in: Liebigs Ann. Chem. , 1986 , pagg. 1281-1308; doi: 10.1002 / jlac.198619860714 .
- ↑ David A. Evans, Stephen W. Kaldor, Todd K. Jones, Jon Clardy, Thomas J. Stout: "Total synthesis of the macrolide antibiotic cytovaricin", in: J. Am. Chem. Soc. , 1990 , 112 , pagg. 7001-7031; doi: 10.1021 / ja00175a038 .
- ↑ James A. Marshall, Richard Sedrani: "Una sintesi convergente, altamente stereoselettiva di una subunità C-11-C-21 dei macbecini", in: J. Org. Chem. , 1991 , 56 , pp. 5496-5498; doi: 10.1021 / jo00019a004 .
- ↑ a b James D. White, Motoji Kawasaki: "Sintesi totale di (+) - latrunculina A", in: J. Am. Chem. Soc. , 1990 , 112 , pagg. 4991-4993; doi: 10.1021 / ja00168a071 .
- ↑ Morris J. Robins, Vicente Samano, Mark D. Johnson: "Composti correlati all'acido nucleico. 58. Ossidazione periodinano, deprotezione primaria selettiva e riduzione notevolmente stereoselettiva dei ribonucleosidi protetti da terz-butildimetilsilile. Sintesi di 9- (β-D-xilofuranosil) adenina o 3'-deuterioadenosina da adenosina ", in: J. Org. Chem. , 1990 , 55 , pp 410-412; doi: 10.1021 / jo00289a004 .
- ^ R. Roger F. Newton, Derek P. Reynolds, Colin F. Webb, Stanley M. Roberts: "Una sintesi totale breve ed efficiente di estere metilico della (±) prostaglandina D 2 che coinvolge un nuovo metodo per la scissione di un dimetil- t-butilsilil etere ", in: J. Chem. Soc., Perkin Trans. 1 , 1981 , pp. 2055-2058; doi: 10.1039 / P19810002055 .
- ↑ Kyriacos C. Nicolaou, RA Daines, TK Chakraborty: "Sintesi totale di amfoteronolide B", in: J. Am. Chem. Soc. , 1987 , 109 , pagg. 2208-2210; doi: 10.1021 / ja00241a063 .
- ↑ Leo A. Paquette, Annette M. Doherty, Christopher M. Rayner: "Sintesi totale di furanocembranolides. 1. Preparazione stereocontrollata di elementi costitutivi eterociclici chiave e assemblaggio di un quadro completo di seco-pseudotteri ", in: J. Am. Chem. Soc. , 1991 , 109 , pagg. 3910-3926; doi: 10.1021 / ja00036a045 .
- ↑ PJ Kocienski: Protecting Groups , p. 40
- ↑ PJ Kocienski: Protecting Groups , pagine 38-39.
- ↑ a b P.J. Kocieński: Protecting Groups , p. 43.
- ↑ JFW McOmie, DE West: 3,3′-Dihyroxybiphenyl In: Organic Syntheses . 49, 1969, pagina 50, doi : 10.15227 / orgsyn.049.0050 ; Coll. Vol.5, 1973, p. 412 ( PDF ).
- ↑ PJ Kocienski: Protecting Groups , pagine 46-49.
- ↑ Yuji Oikawa, Tadao Yoshioka, Osamu Yonemitsu: "Rimozione specifica della protezione o-metossibenzilica mediante ossidazione DDQ", in: Tetrahedron Lett. , 1982 , 23 , pagg. 885-888; doi: 10.1016 / S0040-4039 (00) 86974-9 .
- ↑ Rolf Johansson, Bertil Samuelsson: “Apertura dell'anello riduttivo regioselettivo di 4-metossibenzilidene acetali degli esopiranosidi. Accesso a una nuova strategia di protezione del gruppo. Part 1 ", in: J. Chem. Soc., Perkin Trans. 1 , 1984 , pagg. 2371-2374; doi: 10.1039 / P19840002371 .
- ↑ Letteratura come p- metossibenzile.
- ↑ Michel Bessodes, Dimitri Komiotis, Kostas Antonakis: "Detritilazione rapida e selettiva di alcoli primari utilizzando acido formico", in: Tetrahedron Lett. , 1986 , 27 , pagg. 579-580; doi: 10.1016 / S0040-4039 (00) 84045-9 .
- ↑ B. Helferich: Carbonhydr. Chem. Biochem. , 1948 , 3 , p. 79.
- ↑ ML García, J. Pascual, L. Borràs, JA Andreu, E. Fos, D. Mauleón, G. Carganico, F. Arcamone: "Sintesi di nuovi glicerofosfolipidi eterei strutturalmente correlati al modulatore", in: Tetrahedron , 1991 , 47 , Pp. 10023-10034; doi: 10.1016 / S0040-4020 (01) 96051-X .
- ↑ PJ Kocienski: Protecting Groups , pagine 59-60.
- ↑ PJ Kocienski: Protecting Groups , S. 62nd
- ↑ RE Irlanda, DW Norbeck: "Sintesi convergente di antibiotici ionofori polietere: la sintesi della monensina bis (tetraidrofurano) tramite il riarrangiamento di Claisen di un estere enolato con un gruppo β-uscente", in: J. Am. Chem. Soc. , 1985 , 107 , pagg. 3279-3285; doi: 10.1021 / ja00297a038 .
- ↑ Letteratura vedi allile.
- ^ Paul A. Wender, Carlos RD Correia: "Cyaloadditions diene-diene intramolecolari fotoindotte: un metodo selettivo per la sintesi di anelli complessi a otto membri e polichinani", in: J. Am. Chem. Soc. , 1987 , 109 , pagg. 2523-2525; doi: 10.1021 / ja00242a053 .
- ↑ Elias J. Corey, Mark G. Bock: "Protezione dei gruppi idrossilici primari come eteri metiltiometilici", in: Tetrahedron Lett. , 1975 , 16 , pagg. 3269-3270; doi: 10.1016 / S0040-4039 (00) 91422-9 .
- ↑ Elias J. Corey, Duy H. Hua, Bai Chuan Pan, Steven P. Seitz: "Sintesi totale dell'aplasmomicina", in: J. Am. Chem. Soc. , 1982 , 104 , pagg. 6818-6820; doi: 10.1021 / ja00388a074 .
- ↑ Serge David, Annie Thieffry, Alain Veyrières: "Una procedura delicata per la benzilazione regiospecifica e l'allilazione di composti poliidrossilici tramite i loro derivati stannilenici in solventi non polari", in: J. Chem. Soc., Perkin Trans. 1 , 1981 , Pp. 1796-1801; doi: 10.1039 / P19810001796 .
- ↑ Kaoru Fuji, Shigetoshi Nakano, Eiichi Fujita: "An Improved Method for Methoxymethylation of Alcohols under Mild Acidic Conditions", in: Synthesis , 1975 , pp. 276-277.
- ↑ PJ Kocienski: Protecting Groups , S. 77th
- ↑ H. Nagaoka, W. Rutsch, G. Schmidt, H. Ito, MR Johnson, Y. Kishi: “Sintesi totale delle rifamicine. 1. Sintesi stereocontrollata del blocco alifatico ", in: J. Am. Chem. Soc. , 1980 , 102 , pagg. 7962-7965; doi: 10.1021 / ja00547a037 .
- ^ W. Clark Still, Dominick Mobilio: "Synthesis of asperdiol", in: J. Org. Chem. , 1983 , 48 , pp. 4785-4786; doi: 10.1021 / jo00172a070 .
- ↑ Masahiro Hirama, Mitsuko Uei: "A chiral total synthesis of compactin", in: J. Am. Chem. Soc. , 1982 , 104 , pagg. 4251-4253; doi: 10.1021 / ja00379a037 .
- ↑ W. Clark Still, Shizuaki Murata, Gilbert Revial, Kazuo Yoshihara: "Sintesi del germacranolide citotossico eucannabinolide", in: J. Am. Chem. Soc. , 1983 , 105 , pagg. 625-627; doi: 10.1021 / ja00341a055 .
- ↑ Robert C. Gadwood, Renee M. Lett, Jane E. Wissinger: "Sintesi totale di (±) -poitediol e (±) 4-epipoitediol", in: J. Am. Chem. Soc. , 1984 , 106 , pagg. 3869-3870; doi: 10.1021 / ja00325a032 .
- ↑ Steven D. Burke, Gregory J. Pacofsky: “The ester enolate claisen riarrangiamento. Sintesi totale di (±) -etisolide ", in: Tetrahedron Lett. , 1986 , 27 , pagg. 445-448; doi: 10.1016 / S0040-4039 (00) 85501-X .
- ↑ Toshiyuki Kan, Masaru Hashimoto, Mitsutoshi Yanagiya, Haruhisa Shirahama: "Efficace deprotezione degli alcoli 2- (trimetilsililetossi) metilati (SEM eteri). Sintesi del tirsiferil-23 acetato “, in: Tetrahedron Lett. , 1988 , 29 , pagg. 5417-5418; doi: 10.1016 / S0040-4039 (00) 82883-X .
- ↑ Joseph P. Marino, Scott L. Dax: "Un metodo di desililazione efficiente per la generazione di metidi di o-chinone: applicazione alla sintesi di (+) - e (-) - esaidrocannabinolo", in: J. Org. Chem. , 1984 , 49 , pagg. 3671-3672; doi: 10.1021 / jo00193a051 .
- ↑ Karel F. Bernady, M. Brawner Floyd, John F. Poletto, Martin J. Weiss: “Prostaglandine e congeneri. 20. Sintesi di prostaglandine mediante aggiunta coniugata di reagenti al litio trans-1-alcheniltrialchilalanato. Un nuovo reagente per aggiunte di 1,4 coniugati ", in: J. Org. Chem. , 1979 , 44 , pp. 1438-1447; doi: 10.1021 / jo01323a017 .
- ↑ Elias J. Corey, Haruki Niwa, Jochen Knolle: "Sintesi totale dell'acido (S) -12-idrossi-5,8,14-cis, -10-trans-eicosatetraenoico (Samuelsson's HETE)", in: J. Am . Chem. Soc. , 1978 , 100 , pagg. 1942-1943; doi: 10.1021 / ja00474a058 .
- ^ A b P. Collins, R. Ferrier: Monosacharides - Their Chemistry and their Roles in Natural Products , Wiley, West Sussex 1995, ISBN 0-471-95343-1 .
- ↑ Christopher R. Schmid, Jerry D. Bryant: D- (R) -Glycerinaldehyde Acetonide In: Organic Syntheses . 72, 1995, pagina 6, doi : 10.15227 / orgsyn.072.0006 ; Coll. Vol.9, 1998, p. 450 ( PDF ).
- ↑ András Lipták, János Imre, János Harangi, Pál Nánási, András Neszmélyi: “Idrogenolisi chemio, stereo e regioselettiva dei carboidrati benzilidene acetali. Sintesi di benzil eteri di benzil α-D-, metil β-D-mannopiranosidi e benzil α-D-ramnopiranoside mediante scissione ad anello di derivati del benziliden con il reagente LiAlH 4 -AlCl 3 ", in: Tetrahedron , 1982 , 38 , p. 3721-3727; doi: 10.1016 / 0040-4020 (82) 80083-5 .
- ↑ James A. Marshall, Joseph D. Trometer, Bruce E. Blough, Thomas D. Crute: "Stereohemistry of SN2 'additions to acyclic vinyloxiranes", in J. Org. Chem. , 1988 , 53 , pp. 4274-4282 doi : 10.1021 / jo00253a020
- ↑ T. Tsunoda, M. Suzuki, R. Noyori: "A facile procedure for acetalization under aprotic conditions", in: Tetrahedron Lett. , 1980 , 21 , pagg. 1357-1358; doi: 10.1016 / S0040-4039 (00) 74575-8 .
- ↑ Juji Yoshimura, Shigeomi Horito, Hiroriobu Hashimoto: "Facile Synthesis of 2,3,4,6-Tetra-O-benzyl-D-glucopyranosylidene Acetals Using Trimethylsilyl Trifluoromethanesulfonate Catalyst", in: Chem. Lett. , 1981 , 10 , pagg. 375-376; doi: 10.1246 / cl.1981.375 .
- ↑ Bruce H. Lipshutz, Daniel Pollart, Joseph Monforte, Hiyoshizo Kotsuki: "Pd (II) -catalizzato acetale / chetale idrolisi / reazioni di scambio", in: Tetrahedron Lett. , 1985 , 26 , pagg. 705-708; doi: 10.1016 / S0040-4039 (00) 89114-5 .
- ↑ Kwan Soo Kim, Yang Heon Song, Bong Ho Lee, Chi Sun Hahn: "Scissione efficiente e selettiva di acetali e chetali utilizzando cloruro ferrico adsorbito su gel di silice", in: J. Org. Chem. , 1986 , 51 , p. 404-407; doi: 10.1021 / jo00353a027 .
- ↑ PJ Kocienski: Protecting Groups , pagine 167-170.
- ↑ PJ Kocienski: Protecting Groups , pagine 164-167.
- ↑ PJ Kocienski: Protecting Groups , S. 176th
- ↑ PJ Kocienski: Protecting Groups , pagine 178-180.
- ↑ Samuel J. Danishefsky, Nathan B. Mantlo, Dennis S. Yamashita, Gayle. Schulte: "Percorso conciso alla serie calichemicina-esperamicina: la struttura cristallina di un prototipo di aglicone", in: J. Am. Chem. Soc. , 1988 , 110 , pagg. 6890-6891; doi: 10.1021 / ja00228a051 .
- ↑ John N. Haseltine, Maria Paz Cabal, Nathan B. Mantlo, Nobuharu Iwasawa, Dennis S. Yamashita, Robert S. Coleman, Samuel J. Danishefsky, Gayle K. Schulte: "Sintesi totale del calicheamicinone: nuovi arrangiamenti per l'attivazione del cicloaromatizzazione riduttiva di congeneri agliconi ", in: J. Am. Chem. Soc. , 1991 , 113 , pagg. 3850-3866; doi: 10.1021 / ja00010a030 .
- ↑ PJ Kocienski: Protecting Groups , S. 119th
- ↑ Satomi Niwayama: "Monoidrolisi selettiva altamente efficiente dei diesteri simmetrici", in: J. Org. Chem. , 2000 , 65 , pp. 5834-5836; doi: 10.1021 / jo0001986 .
- ↑ JM Khurana, Arti Sehgal: "Una procedura efficace e conveniente per l'idrolisi degli esteri", in: Org. Prep. Proced. Ind. , 1994 , 26 , pagg. 580-583.
- ^ Robert V. Stevens, Albert WM Lee: "Stereochimica della reazione di Robinson-Schoepf. Una sintesi stereospecifica totale degli alcaloidi di difesa della coccinella precoccinellina e coccinellina ”, in: J. Am. Chem. Soc. , 1979 , 101 , pagg. 7032-7035; doi: 10.1021 / ja00517a042 .
- ↑ J. Wrobel, K. Takahashi, V. Honkan, G. Lannoye, JM Cook, Steven H. Bertz: "α-Lithio chetones. 1. Sintesi stereocontrollata del (±) -modhephene tramite la reazione di Weiss ", in: J. Org. Chem. , 1983 , 48 , pp. 139-141; doi: 10.1021 / jo00149a034 .
- ↑ Dennis D. Keith, John A. Tortora, Roxana Yang: "Synthesis of L-2-amino-4-methoxy-trans-but-3-enoic acid", in: J. Org. Chem. , 1978 , 43 , Pp. 3711-3713; doi: 10.1021 / jo00413a016 .
- ↑ Peter Mohr, Nada Waespe-Šarčević, Christoph Tamm, Krystyna Gawronska, Jacek K. Gawronski: "A Study of Stereoselective Hydrolysis of Symmetrical Diester with Pig Liver Esterase", in: Helv. Chim. Acta , 1983 , 66 , pagg. 2501-2511; doi: 10.1002 / hlca.19830660815 .
- ↑ Théophile Tschamber, Nada Waespe-Šarčević, Christoph Tamm: "Sintesi stereocontrollata di un epimero del segmento C (19) -to-C (27) della rifamicina S", in: Helv. Chim. Acta , 1986 , 69 , pagg. 621-625; doi: 10.1002 / hlca.19860690311 .
- ↑ Yves Rubin, Carolyn B. Knobler, Francois Diederich: "Precursori dei ciclo [n] atomi di carbonio: da 3,4-dialchinil-3-ciclobutene-1,2-dioni e 3,4-dialchinil-3-ciclobutene-1 , 2-dioli a ciclobutenodeidroannuleni e ossidi superiori di carbonio ", in: J. Am. Chem. Soc. , 1990 , 112 , pagg. 1607-1617; doi: 10.1021 / ja00160a047 .
- ↑ Sunggak Kim, Yong Gil Kim, Deog-il Kim: "Un nuovo metodo per la diossolanizzazione selettiva dei chetoni in presenza di aldeidi", in: Tetrahedron Lett. , 1992 , 33 , pagg. 2565-2566; doi: 10.1016 / S0040-4039 (00) 92243-3 .
- ↑ G. Bauduin, D. Bondon, Y. Pietrasanta, B. Pucci: "Reactions de transcetalisation - II: Influence des facteurs steriques et electroniques sur les energies de cetalisation", in: Tetrahedron , 1978 , 34 , pp. 3269-3274 ; doi: 10.1016 / 0040-4020 (78) 80243-9 .
- ↑ John E. McMurry, Stephen J. Isser: "Total synthesis of longifolene", in: J. Am. Chem. Soc. , 1972 , 94 , pagg. 7132-7137; doi: 10.1021 / ja00775a044 .
- ^ MP Bosch, M. Pilar Bosch, Francisco Camps, Jose Coll, Angel Guerrero, Toshio Tatsuoka, Jerrold Meinwald: "A stereoselective total synthesis of (±) -muzigadial", in: J. Org. Chem. , 1986 , 51 , Pp. 773-784; doi: 10.1021 / jo00356a002 .
- ↑ Ulrich Schmidt, Thomas Beuttler, Albrecht Lieberknecht, Helmut Griesser: "Aminoacidi e peptidi - XXXXII. Sintesi di Chlamydocin + epi-Chlamydocin “, in: Tetrahedron Lett. , 1983 , 24 , pagg. 3573-3576; doi: 10.1016 / S0040-4039 (00) 88171-X .
- ↑ Elias J. Corey, Plato A. Magriotis: "Sintesi totale e configurazione assoluta di 7,20-diisocyanoadociane", in: J. Am. Chem. Soc. , 1987 , 109 , pagg. 287-289; doi: 10.1021 / ja00235a052 .
- ↑ Elias J. Corey, Kyriacos C. Nicolaou, Takeshi Toru: "Sintesi totale di (±) -vermiculine", in: J. Am. Chem. Soc. , 1975 , 97 , pagg. 2287-2288; doi: 10.1021 / ja00841a058 .
- ↑ Tainejiro Hiyama, Akihiro Kanakura, Hajime Yamamoto, Hitosi Nozaki: “Via generale verso α, aldeidi β-insature di struttura omoterpenoide e terpenoide. Sythesis of JH-II and β-Sinensal ", in: Tetrahedron Lett. , 1978 , 19 , pagg. 3051-3054; doi: 10.1016 / S0040-4039 (01) 94936-6 .
- ↑ F. Huet, A. Lechevallier, M. Pellet, JM Conia: “Wet Silica Gel; A Convenient Reagent for Deacetalization ", in: Synthesis , 1978 , pp. 63-64.
- ↑ F.Zymalkokowski: Catalytic Hydrogenation , Ferdinand Enke Verlag, Stoccarda 1965, pp. 127-133.
- ↑ PJ Kocienski: Protecting Groups , S. 136th
- ↑ PJ Kocienski: Protecting Groups , pagine 139-142.
- ↑ Ahmed M. Tafesh, Jens Weiguny: "A Review of the Selective Catalytic Reduction of Aromatic Nitro Compounds into Aromatic Amines, Isocyanates, Carbamates, and Ureas Using CO", in: Chem. Rev. , 1996 , 96 , p. 2035– 2052; doi: 10.1021 / cr950083f .
- ↑ Evan L. Allred, Boyd R. Beck, Kent J. Voorhees: "Formazione di doppi legami carbonio-carbonio mediante reazione di dialogenuri vicinali con sodio in ammoniaca", in: J. Org. Chem. , 1974 , 39 , p. 1426-1427; doi: 10.1021 / jo00926a024 .
- ↑ Timothy S. Butcher, Feng Zhou, Michael R. Detty: “Debrominations of vic-Dibromides with Diorganotellurides. 1. Stereoselettività, tassi relativi e implicazioni meccanicistiche ", in: J. Org. Chem. , 1998 , 63 , pp. 169-176; doi: 10.1021 / jo9713363 .
- ^ CJ Li, David N. Harpp: "Bis (trifeniltanil) tellururo un reagente delicato e selettivo per la tellurazione e la debrominazione", in: Tetrahedron Lett. , 1990 , 31 , pagg. 6291-6293; doi: 10.1016 / S0040-4039 (00) 97045-X .
- ↑ Corrado Malanga, Serena Mannucci, Luciano Lardicci: “Attivazione del legame carbonio-alogeno mediante catalizzatore di nichel: sintesi di alcheni, da 1,2-dialogenuri”, in: Tetrahedron , 1998 , 54 , pp. 1021-1028; doi: 10.1016 / S0040-4020 (97) 10203-4 .
- ↑ Byung Woo Yoo, Seo Hee Kim, Jun Ho Kim: "A Mild, Efficient, and Selective Debromination of vic-Dibromides to Alkenes with Cp 2 TiCl 2 / Ga System", in: Bull. Korean Chem. Soc. , 2010 , 31 , pagg. 2757-2758; doi: 10.5012 / bkcs.2010.31.10.2757 .
- ^ Antonius JH Klunder, Jie Zhu, Binne Zwanenburg: "The Concept of Transient Chirality in the Stereoselective Synthesis of Functionalized Cycloalkenes Applying the Retro-Diels-Alder Methodology", in: Chem. Rev. , 1999 , 99 , pp. 1163-1190 ; doi: 10.1021 / cr9803840 .
- ↑ Hideyuki Tanaka, Takashi Kamikubo, Naoyuki Yoshida, Hideki Sakagami, Takahiko Taniguchi, Kunio Ogasawara: "Enantio- and Diastereocontrolled Synthesis of (-) - Iridolactone and (+) - Pedicularis-lactone", in: Org. Lett. , 2001 , 3 , pagg. 679-681; doi: 10.1021 / ol0070029 .
- ↑ Martin Banwell, David Hockless, Bevyn Jarrott, Brian Kelly, Andrew Knill, Robert Longmore, Gregory Simpson: "Approcci chemioenzimatici ai nuclei decaidro-come-indacene associati alla classe degli insetticidi spinosyn", in: J. Chem. Soc. , Perkin Trans. 1 , 2000 , pagg. 3555-3558; doi: 10.1039 / b006759h .
- ↑ Wenzel E. Davidsohn, Malcolm C. Henry: "Organometallic Acetylenes of the Main Groups III-V", in: Chem. Rev. , 1967 , 67 , pp. 73-106; doi: 10.1021 / cr60245a003 .
- ↑ Barry J. Teobald: "La reazione di Nicholas: l'uso di cationi propargilici stabilizzati con dicobalto in sintesi", in: Tetrahedron , 2002 , 58 , pp. 4133-4170; doi: 10.1016 / S0040-4020 (02) 00315-0 .
- ↑ Kenneth M. Nicholas, R. Pettit: "An alkyne protection group", in: Tetrahedron Lett. , 1971 , 37 , pagg. 3475-3478; doi: 10.1016 / S0040-4039 (01) 97209-0 .
- ↑ Richard E. Connor, Kenneth M. Nicholas: "Isolamento, caratterizzazione e stabilità degli ioni α - [(etinil) dicobalto esacarbonile] carbonio", in: J. Organomet. Chem. , 1977 , 125 , C45-C48; doi: 10.1016 / S0022-328X (00) 89454-1 .
- ↑ Rosa F. Lockwood, Kenneth M. Nicholas: “Gli ioni carbenio stabilizzati con metallo di transizione come intermedi sintetici. I. ioni α - [(alchinil) dicobalto esacarbonil] carbenio come agenti propargilanti ", in: Tetrahedron Lett. , 1977 , pagg. 4163-4166; doi: 10.1016 / S0040-4039 (01) 83455-9 .
- ^ KM Nicholas, R. Pettit: "Sulla stabilità degli ioni α- (alchinil) dicobalto esacarbonilcarbonio", in: J. Organomet. Chem. , 1972 , 44 , C21-C24; doi: 10.1016 / 0022-328X (72) 80037-8 .
- ↑ T. Reichstein, A. Grüssner: "Una sintesi produttiva dell'acido L-ascorbico (vitamina C)", in: Helv. Chim. Acta , 1934 , 17 , pagg. 311-328; doi: 10.1002 / hlca.19340170136 .
- ^ KC Nicolaou, EJ Sorensen: Classics in Total Synthesis: Targets, Strategies, Methods , VCH Verlagsgesellschaft mbH, Weinheim 1996, pp. 711-729, ISBN 3-527-29284-5 .
- ↑ Peter GM Wuts, Theodora W. Greene: Green's Protective Groups in Organic Synthesis , 4a edizione, John Wiley & Sons Inc., Hoboken, New Jersey, pp. 10-13; ISBN 0-471-69754-0 .
- ↑ JM McClure, Samuel J. Danishefsky: "Una nuova reazione di arilazione Heck: accesso rapido ai congeneri di FR 900482", in: J. Am. Chem. Soc. , 1993 , 115 , pagg. 6094-6100; doi: 10.1021 / ja00067a026 .
- ↑ Serge L. Beaucage, Radhakrishman P. Iyer: "Advances in the Synthesis of Oligonucleotides by the Phosphoramidite Approach", in: Tetrahedron , 1992 , 48 , pp. 2223-2311; doi: 10.1016 / S0040-4020 (01) 88752-4 .
- ↑ Michael Schelhaas, Herbert Waldmann: "Strategie del gruppo di protezione nella sintesi organica", in: Angewandte Chemie , 1996 , 103 , 2194; doi: 10.1002 / anie.19961081805 .
- ↑ Marco Eissen, Radoslaw Mazur, Heinz-Georg Quebbemann e Karl-Heinz Pennemann: "Atom Economy and Yield of Synthesis Sequences", in: Helv. Chim. Acta , 2004 , 87 , pagg. 524-535; doi: 10.1002 / hlca.200490050 .
- ^ Marco Eissen, Jürgen O. Metzger: "Environmental Performance Metrics for Daily Use in Synthetic Chemistry", in: Chemistry - A European Journal , 2002 , 8 , pp. 3580-3585; doi: 10.1002 / mento.200247273 .
- ↑ Marco Eissen, Jürgen O. Metzger, Eberhard Schmidt, Uwe Schneidewind: 10 Years After "Rio" - Concetti per il contributo della chimica allo sviluppo sostenibile , in: Angewandte Chemie , 2002 , 114 , pp. 402–425; doi : 10.1002 / 1521-3757 (20020201) 114: 3 <402 :: AID-ANGE402> 3.0.CO; 2-D .
- ^ Siegfried Hauptmann : Organische Chemie , 2a edizione, VEB Deutscher Verlag für Grundstoffindustrie, Lipsia 1985, ISBN 3-342-00280-8 , pp. 621-622.
- ↑ Hideaki Muratake, Harumi Kumagami, Mitsutaka Natsume: studi sintetici sugli alcaloidi marini hapalindoli. Parte 3 Sintesi totale degli (±) -hapalindoli H e U , in: Tetrahedron , 1990 , 46 , pp. 6351-6360; doi: 10.1016 / S0040-4020 (01) 96007-7 .
- ↑ Hideaki Muratake, Mitsutaka Natsume: studi sintetici di alcaloidi marini hapalindoli. Parte I Sintesi totale degli (±) -hapalindoli J e M , in: Tetrahedron , 1990 , 46 , pp. 6331-6342; doi: 10.1016 / S0040-4020 (01) 96005-3 .
- ↑ Hideaki Muratake, Mitsutaka Natsume: studi sintetici di alcaloidi marini hapalindoli. Parte 2. Riduzione del litio-alluminio idruro del doppio legame carbonio-carbonio ricco di elettroni coniugato con il nucleo dell'indolo , in: Tetrahedron , 1990 , 46 , pp. 6343-6350; doi: 10.1016 / S0040-4020 (01) 96006-5 .
- ↑ Phil S. Baran, Thomas J. Maimone, Jeremy M. Richter: Sintesi totale di prodotti naturali marini senza l'utilizzo di gruppi protettivi , in: Nature , 446 , pp. 404-408; doi: 10.1038 / nature05569 .