
Spettroscopia ad infrarossi
La spettroscopia infrarossa , spettroscopia IR corta e fino agli anni '60, spettroscopia infrarossa chiamata, è un metodo fisico di analisi che funziona con la radiazione infrarossa (lunghezza d'onda: da 800 nm a 1 mm). Il metodo appartiene ai metodi della spettroscopia molecolare , che si basano sull'eccitazione degli stati energetici nelle molecole.

La spettroscopia IR viene utilizzata per la determinazione quantitativa di sostanze note, identificate mediante uno spettro di riferimento, o per la delucidazione strutturale di sostanze sconosciute.
Metodi spettroscopici molecolari simili sono la spettroscopia Raman , che fornisce anche informazioni sulle vibrazioni nella gamma dell'infrarosso, e la spettroscopia UV/VIS nella gamma di frequenze più alte.
Al giorno d'oggi, vengono utilizzati principalmente spettrometri a infrarossi a trasformata di Fourier (spettrometri FTIR). Rispetto agli spettrometri a dispersione utilizzati in passato, questi offrono una serie di vantaggi decisivi. Ad esempio, gli spettrometri FTIR hanno un rapporto segnale-rumore significativamente più elevato e richiedono tempi di misurazione significativamente più brevi. I risultati della spettroscopia a infrarossi sono mostrati nella spettrografia a infrarossi .
varianti
Da un punto di vista spettroscopico, si distingue tra il vicino infrarosso (NIR; numero d'onda : 12500–4000 cm -1 ; lunghezza d'onda: 0,8–2,5 µm, vedi spettroscopia del vicino infrarosso ), l' infrarosso medio o classico (normale) ( MIR; numero d'onda: 4000 –400 cm −1 lunghezza d'onda: 2,5–25 µm) e il lontano infrarosso (FIR; numero d'onda: 400–10 cm −1 ; lunghezza d'onda: 25–1000 µm), poiché si possono osservare diversi fenomeni a causa all'assorbimento nella rispettiva area sono. Un assorbimento nella gamma FIR porta principalmente alla rotazione di intere molecole. Nella gamma MIR e nella gamma NIR, l'oscillazione di atomi o gruppi di atomi è eccitata ai loro legami molecolari, con le cosiddette armoniche della gamma MIR rilevabili nella gamma NIR (in particolare dei legami CH, OH e NH).
La spettroscopia nel medio infrarosso - spesso indicata semplicemente come spettroscopia IR - è una tecnica potente nell'analisi chimica delle sostanze organiche. Consente di formulare affermazioni dirette sulla presenza ed eventualmente sulla concentrazione di gruppi funzionali attivi nell'infrarosso . La spettroscopia nel vicino infrarosso (NIRS) è utilizzata (spesso, specialmente nelle applicazioni per la revisione rapida di analisi di processo di sostanze e miscele ), ma ai modelli chemiometrici computerizzati si basano con accompagnamento, analisi di riferimento casualmente errate.
Negli spettri IR, a differenza degli spettri UV , spesso non è l' assorbimento ma piuttosto la trasmissione che viene utilizzata come misura della permeabilità della radiazione di eccitazione. La trasmissione è tracciata sull'asse verticale, aumentando verso l'alto - le aree di bassa trasmissione della radiazione IR provocano una deflessione verso il basso. Tuttavia, viene utilizzata anche la rappresentazione come spettro di assorbimento, in particolare con la tecnologia ATR.
Principio di misura
Assorbimento delle radiazioni IR
Quando una sostanza viene irradiata con onde elettromagnetiche , vengono assorbite determinate gamme di frequenza. In termini di energia, la radiazione infrarossa risiede nell'intervallo dei livelli di rotazione di piccole molecole e dei livelli di vibrazione dei legami molecolari, ad es. Cioè, l'assorbimento fa vibrare i legami. Sono visibili sotto forma di deviazioni nello spettro misurato (diagramma). Poiché le energie o frequenze necessarie per questo sono caratteristiche dei rispettivi legami, anche i materiali possono essere identificati in questo modo. La spettroscopia IR è quindi chiarificatrice della struttura.
L'interazione tra la radiazione elettromagnetica e la molecola può avvenire solo se c'è una carica elettrica in movimento nella molecola. Questo è sempre il caso quando la molecola ha un momento di dipolo variabile o inducibile (IR-attivo). Nelle molecole con vibrazioni simmetriche al centro di simmetria, non ci sono variazioni del momento di dipolo (IR inattivo). Tali vibrazioni "proibite" sono spesso attive Raman .
Il caso più semplice è una molecola biatomica. Nel caso di molecole poliatomiche, le vibrazioni fondamentali sono sovrapposte. Di conseguenza, si vedono una serie di bande di assorbimento che devono essere interpretate.
Modello meccanico


Il modello più semplice concepibile che può essere utilizzato per spiegare le vibrazioni e le eccitazioni di rotazione è il modello classico dell'interazione di un dipolo elettrico permanente in un campo elettromagnetico. Successivamente, modelli migliori, come il modello della meccanica quantistica, possono anche descrivere molecole senza un momento di dipolo permanente.
Esistono interazioni attrattive e repulsive tra gli atomi e i loro vicini. Pertanto, la distanza di legame ottimale nella molecola è nel minimo della funzione potenziale. Meccanicamente, questo può essere immaginato come se gli atomi fossero collegati tra loro da molle. La forza necessaria per deflettere una molla è descritta dalla legge delle molle di Hooke . Se si porta una tale molecola in un campo elettrico, come esiste in un condensatore a piastre, per esempio, la molecola prima si allineerà con il suo momento di dipolo lungo il campo elettrico e in secondo luogo aumenterà la sua distanza di legame. Se ora viene applicata una tensione alternata o se la molecola viene eccitata con un'onda elettromagnetica, i gruppi funzionali “appesi” ai legami iniziano a vibrare e ruotare. Il modello meccanicistico è tuttavia di utilità limitata, poiché non spiega, ad esempio, perché per l'eccitazione IR siano consentite solo energie discrete e perché anche le molecole senza un momento di dipolo permanente mostrino assorbimento IR.
Modello meccanico quantistico

Come nel modello della meccanica classica, la base del modello quantomeccanico della vibrazione e dell'eccitazione rotatoria è la funzione potenziale. Il minimo della funzione potenziale può essere approssimato da una parabola. Tale parabola risulta dall'integrazione della legge delle molle di Hooke.
Se una molecola è ora eccitata con radiazione elettromagnetica, i legami di valenza all'interno della molecola possono essere impostati in oscillazioni e rotazioni se l'energia è sufficiente per sollevarla dal suo stato base al primo stato di oscillazione eccitato. Per determinare questa energia, l'equazione di Schrödinger deve essere risolta per questo potenziale. Dopo aver separato il movimento relativo dei nuclei atomici e degli elettroni ( approssimazione di Born-Oppenheimer ), la soluzione dell'equazione di Schrödinger risulta in una relazione tra l'energia richiesta, la forza di legame ( k ) e la massa ridotta ( μ ). A differenza dell'oscillatore armonico classico, nel caso della meccanica quantistica l'energia di vibrazione è quantizzata dal numero quantico di vibrazione v .
Equazione di Schrödinger:

Soluzione dell'equazione di Schrödinger:

forme d'onda
Nelle sostanze inorganiche e organiche, le vibrazioni meccaniche si verificano quando viene assorbita la radiazione della gamma infrarossa. Secondo R. Mecke , le vibrazioni molecolari possono essere suddivise in due tipi:
- Allungamento vibrazioni (allungare vibrazioni): vibrazioni lungo l'asse legame di due atomi o parti di una molecola ad allungamento o compressione del legame
-
Vibrazioni di deformazione : vibrazioni sotto la deformazione dell'angolo di legame
- nel piano (vibrazioni di flessione/flessione):
- fuori piano (vibrazioni torsionali/inclinabili) - per lo più perpendicolari al piano del legame
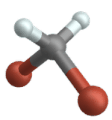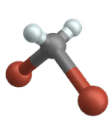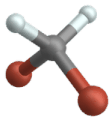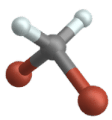
Stiramento simmetrico
(Engl. Simmetrico allungamento )
Allungamento antisimmetrico
( ingl . Allungamento antisimmetrico )
Vibrazione di flessione
(inglese a forbice o flessione )
Vibrazione oscillante
(Inglese Rocking )
Oscillazione a dondolo
(engl. Wagging )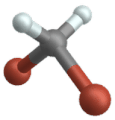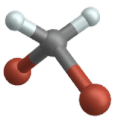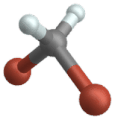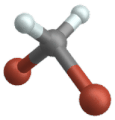
Vibrazioni torsionali
(inglese torsione o torsing )






Poiché le vibrazioni molecolari di alcuni gruppi atomici nell'intervallo 4000-1500 cm -1 sono particolarmente caratteristiche, la spettroscopia IR è adatta per determinare alcuni elementi strutturali, in particolare i gruppi funzionali della molecola in esame.
Spettro di vibrazione rotazionale
Lo spettro rotazione-oscillazione ideale

L'acido cloridrico (HCl) è una molecola biatomica con un momento di dipolo pronunciato. In termini semplificati , la molecola di HCl può essere vista come un top lineare . Come nella spettroscopia di rotazione pura , il modello del rotatore rigido può essere utilizzato per descrivere gli spettri. Per una molecola lineare la regola di selezione speciale per una transizione rotazionale è:
- ,

dove rappresenta il numero quantico rotazionale. Se il momento di dipolo è parallelo all'asse di rotazione principale (come nel caso dell'ammoniaca , ad esempio ), sarebbe anche possibile. La regola di selezione speciale per una transizione di oscillazione dell'oscillatore armonico è:



Le transizioni tra i diversi livelli rotazionali ( ) dello stato vibrazionale fondamentale ( ) e i livelli rotazionali ( ) dello stato vibrazionale eccitato ( ) possono essere utilizzati in due gruppi, il ramo R (destra) per le transizioni e il ramo P (sinistra ) per le transizioni , divise sono e sono abbozzate in figura. Poiché HCl non è consentito, non viene visualizzato alcun ramo Q. Lo schema del livello di energia è mostrato nella figura sopra. La lunghezza della freccia mostra chiaramente che ogni transizione è un apporto energetico di






richiede, dove è la costante di rotazione con l'unità . Questo include il momento d' inerzia della molecola rispetto all'asse di rotazione.



Quanto segue si applica alla differenza di energia alla transizione vicina

Pertanto, nello spettro di vibrazione rotazionale ideale (pannello inferiore) i picchi hanno una distanza costante di 2 B . Nell'esperimento, i picchi sono distanziati in modo leggermente diverso perché il modello non:
- l'anarmonia della vibrazione
- la distanza atomica espansa attraverso le forze centrifughe (approssimazione: rotatore rigido )
- e si osserva l'interazione del movimento vibratorio e rotatorio.
Il vero spettro di rotazione-oscillazione
Lo spettro di rotazione-oscillazione reale si discosta dall'ideale a causa dei limiti dei modelli utilizzati. Le distanze tra i picchi (deflessione in ampiezza nel diagramma, dette anche bande ) non sono esattamente 2 B , come nel caso ideale , ma diventano minori all'aumentare del numero d'onda nel ramo R, ma maggiori nel ramo P a causa della interazione di rotazione e oscillazione. Il diagramma a lato mostra uno spettro registrato di acido cloridrico in fase gassosa. Nella struttura fine dello spettro si nota chiaramente un raddoppio di tutte le bande dovuto ai due isotopi diversamente inerti del cloro.
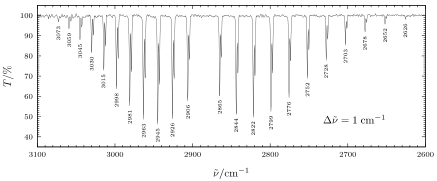
Spettro di vibrazione rotazionale di acido cloridrico gassoso a temperatura ambiente.

Spettro di vibrazione rotazionale dell'anidride carbonica gassosa misurato a temperatura ambiente con diverse risoluzioni.


Posizione e valutazione delle bande di assorbimento IR
Legami chimici più forti e atomi di massa più piccola causano massimi di assorbimento nello spettro IR a grandi numeri d'onda (alta energia), mentre grandi masse causano massimi di assorbimento IR a piccoli numeri d'onda (bassa energia) (vedi deuterazione ). L'energia è proporzionale al quadrato del momento di dipolo permanente. Pertanto, le molecole polari forniscono intense transizioni rotazionali. Tuttavia, se si confrontano tra loro le ampiezze dei picchi di una singola molecola, si nota che la forza delle transizioni inizialmente aumenta rapidamente all'aumentare di J , passa per un massimo e infine decresce nuovamente per J grande . La ragione di ciò è che la forza riflette le degenerazioni dei vari stati rotazionali e i numeri di occupazione dei livelli rotazionali nello stato iniziale. Il grado di degenerazione aumenta con l'aumentare di J , che porta ad un'energia più elevata. D'altra parte, i numeri di occupazione diminuiscono con l'aumento dell'energia, il che alla fine porta a una diminuzione dell'intensità della radiazione.

Gli spettri IR vengono interpretati in modo da poter derivare la forma molecolare dalla curva dello spettro IR misurato . Vengono misurate le diverse varianti di oscillazione delle molecole e si possono trarre conclusioni sulla presenza di determinati elementi strutturali. Un tipico spettro IR va da 4000 cm -1 a 400 cm -1 ( numero d'onda ). Il numero d'onda è dato nell'unità alquanto insolita cm −1 . Tuttavia, questo ha il vantaggio che il numero d'onda e l'energia di eccitazione sono proporzionali. Il numero d'onda moltiplicato per il quanto di Planck e la velocità della luce dà l'energia. In uno spettro IR, ogni molecola lascia un tipico schema di bande di assorbimento. La lunghezza di queste bande di solito non viene valutata numericamente, ma valutata solo con attributi come " forte " o " non molto intenso ". Da circa 1500 cm -1 in giù, l'assegnazione delle singole bande a determinate vibrazioni molecolari diventa significativamente più difficile, poiché il numero di possibili sovrapposizioni dovute alle vibrazioni della struttura diventa molto elevato. Questa cosiddetta area dell'impronta digitale dovrebbe idealmente fornire uno spettro caratteristico per ogni tipo di molecola. Si può infatti ipotizzare che alcune differenze non possano essere rappresentate dall'accuratezza di misura dell'analisi spettrale. In quest'area, però, sono presenti anche alcune fasce che sono caratteristiche di alcuni elementi strutturali. Le coppie di enantiomeri mostrano spettri completamente identici a meno che non venga utilizzata luce IR polarizzata linearmente . Se non sono presenti sostanze pure ma miscele , lo spettro IR contiene informazioni sugli elementi strutturali di tutti i componenti della miscela. Con spettri ad alta risoluzione corrispondentemente, tuttavia, i singoli componenti possono ancora essere rilevati sulla base di assorbimenti caratteristici di determinati gruppi di sostanze o di una combinazione di più assorbimenti. Spesso si fa un confronto diretto con uno spettro della sostanza pura.


L'assegnazione individuale delle bande si basa su dati empirici tabulati. La posizione esatta di una banda può essere spostata a numeri d'onda superiori o inferiori a causa dell'ambiente chimico del rispettivo gruppo atomico, il che consente di trarre ulteriori conclusioni sulla vicinanza di determinati elementi strutturali. Quando si interpretano le posizioni delle bande, tuttavia, si deve tener conto che vari effetti ottici possono anche portare a spostamenti di banda, ad es. B. effetti di interferenza e di allineamento nonché la polarizzazione della materia da parte della luce. Ciò rende difficile interpretare tali cambiamenti.
| Denominazione secondo Mecke / gruppo atomico | Intervallo di numeri d'onda in cm −1 |
commento |
|---|---|---|
| (–C – H x ) | 2850-3200 | ν = vibrazione di allungamento; × = 1… 3, numero di atomi di idrogeno legati |
| (–C – H x ) | 1400 | δ = vibrazione di deformazione; × = 1… 3, numero di atomi di idrogeno legati |
| -C = C | 1650 | |
| –C≡C | 2200-2500 | |
| (-OH) | 3200-3600 | In relazione ai legami idrogeno, bande spesso su un ampio intervallo spettrale, ad esempio nel caso dell'acqua liquida |
| (-OH) | 2500-3000 | nei gruppi carbossilici |
| -C = O | 1700 | |
| (–C≡N) | 2200-2260 | Vibrazione di allungamento, ad es. B. in ABS o SAN (vedi foto) |
| (–NH) | 3100-3500 | |
| -NO 2 | 1500 | |
| -C-X | <1500 | dove X è un alogeno è |
| ν come (O = C = O) | 2349 | Diossido di carbonio. |
Tecniche di spettroscopia IR
trasmissione
Uno dei metodi più comunemente usati è misurare la trasmittanza di un campione. A tale scopo, un campione viene irradiato con infrarossi e viene misurata la proporzione della radiazione di misura che è penetrata nel campione. Se il campione ha legami IR attivi o gruppi di molecole, nello spettro di trasmissione si possono vedere aree di minore intensità (bande di assorbimento). Secondo la legge di Lambert-Beer (tra l'altro), l' intensità della banda aumenta con lo spessore del campione (si noti, tuttavia, che questa legge generalmente si applica solo approssimativamente perché non è compatibile con le equazioni di Maxwell La tecnologia di misura più o deviazioni meno grandi. Spiegazioni dettagliate e possibili soluzioni possono essere trovate in). Inoltre, viene riflessa anche una parte della radiazione ; parti corrispondenti di solito hanno l'effetto di una maggiore trasmissione ridotta sull'intero spettro e possono essere in gran parte rimosse con una correzione della linea di base.
A seconda della forma del campione (gassoso, liquido, solido) esistono particolari tecniche di misura che si differenziano per il tipo di preparazione del campione e le celle di misura (portacampione con percorsi ottici più o meno complicati). La disposizione di misurazione più semplice corrisponde a un semplice supporto per campioni che fissa un solido (ad esempio una lamina o una lastra) nel percorso del raggio dello spettrometro. Nei campioni con interfacce parallele (come un film), invece, le interferenze derivano da riflessioni multiple nel campione (vedi anche anelli newtoniani ) che, a seconda dello spessore e del parallelismo piano, falsificano lo spettro, ma le interferenze possono essere utilizzate anche per determinare lo spessore dello strato .


Una tecnica ampiamente utilizzata per misurare campioni solidi consiste nell'incorporare il corpo solido in un materiale di supporto trasparente IR (noto anche come matrice). Inorganici sali quali alogenuri dei metalli alcalini sono principalmente utilizzati ; una matrice comune è bromuro di potassio (KBr). Per misurare i solidi, la polvere secca di bromuro di potassio (KBr è igroscopica ) viene miscelata con una sostanza campione e macinata insieme intensamente. Questa miscela viene quindi pressata in una compressa in un cilindro di acciaio sotto vuoto e ad alta pressione meccanica. A questa pressione, il bromuro di potassio diventa plastico e il flusso freddo (di solito anche otticamente) crea “pellet” trasparenti. Questi possono quindi essere semplicemente posizionati nel percorso del raggio.
I campioni liquidi possono essere collocati tra due lastre trasparenti agli infrarossi realizzate con cristalli singoli di alogenuri di metalli alcalini o, raramente, lastre polimeriche trasparenti. Questo può essere fatto staticamente o dinamicamente sotto forma di un'unità di flusso. I campioni gassosi sono generalmente posti in cuvette speciali .
Misura dello spessore dello strato

Film in polipropilene con interferenze (spessore circa 38 µm)
|

Poiché c'è interferenza con campioni sottili piano-parallelo, lo spettro effettivo è sovrapposto da un treno d'onda sinusoidale. Lo spessore del film può essere calcolato dalla posizione dei massimi.

con l' indice di rifrazione del campione e il numero di massimi nell'intervallo spettrale tra i numeri d'onda e .




È possibile eliminare l'interferenza nelle lamine piegandole o irruvidindole. In tal modo, tuttavia, viene aumentata la percentuale di riflessione diffusa. Un metodo alternativo è la cosiddetta analisi della dispersione. Lo spettro misurato è modellato sulla base dell'ottica delle onde e della teoria della dispersione e si ottengono le costanti ottiche, lo spessore dello strato e i parametri dell'oscillatore. In questo modo è generalmente possibile eliminare un buon numero di effetti ottici.
Effetto Christiansen
L' effetto Christiansen (secondo Christian Christiansen ) è un artefatto nelle misurazioni della trasmissione IR di sostanze legate nel bromuro di potassio o in un'altra matrice (i cosiddetti pellet). Si manifesta principalmente attraverso bande asimmetriche nello spettro. Il bordo ad alta frequenza mostra un andamento molto ripido (spesso più alto della base dello spettro). Il fianco a bassa frequenza, invece, tende a mostrare un andamento piatto e allungato. La causa risiede nella dispersione anomala in prossimità di forti bande di assorbimento . Tuttavia, l'effetto si osserva solo in pellet in cui la sostanza da esaminare è a grana troppo grossa, o in campioni con una maggiore differenza nell'indice di rifrazione tra la sostanza da esaminare e il materiale di matrice. Ciò significa che l'effetto può essere ridotto al minimo o impedito se la sostanza viene ridotta di dimensioni mediante macinazione.
Riflessione e assorbimento



Un altro metodo è la riflessione (esterna) della radiazione IR su una superficie liscia. Il metodo sfrutta il fatto che il grado di riflessione di un campione dipende dal complesso indice di rifrazione del campione (cfr. formule di Fresnel ). Nell'area dei centri di oscillazione, aumenta l'assorbimento, che è rappresentato nell'indice di rifrazione complesso da un aumento del coefficiente di estinzione (la parte immaginaria dell'indice di rifrazione). Questo aumento è accompagnato da una variazione dell'indice di rifrazione attorno alla banda di assorbimento, che è in prima approssimazione proporzionale a tale aumento. Sia l'aumento dell'indice di rifrazione che quello dell'indice di assorbimento portano ad un aumento della riflessione del campione per questa gamma di frequenze e una banda nello spettro di riflessione. Per oscillatori deboli, come quelli che si trovano principalmente nei materiali organici, predomina l'influenza della variazione dell'indice di rifrazione, motivo per cui gli spettri mostrano forme di banda simili a derivati , mentre l'influenza dell'indice di assorbimento aumenta con l'aumentare della forza dell'oscillatore e determina la banda forma. La ragione delle forme a banda derivate risiede nella dispersione anomala in prossimità dei centri di assorbimento di un materiale, cioè l'indice di rifrazione aumenta con la lunghezza d'onda invece di diminuire come al solito. Questo porta ad una riflettività modificata e quindi alle bande distorte osservate. Una crescente influenza dell'indice di assorbimento, invece, porta a forme di banda che temporaneamente assomigliano a bande di assorbimento, prima che diventino bande molto larghe con plateau in prossimità del 100% di riflessione, il cui abbassamento indica poi la presenza di oscillatori più deboli .
La tecnica è adatta, tra l'altro, per misurare campioni fortemente assorbenti. Lo svantaggio principale del metodo è che gli spettri di riflessione differiscono notevolmente dagli spettri di trasmissione. Recentemente è stato proposto un nuovo metodo di valutazione quantitativa per gli spettri di riflessione dei materiali organici, che è una valutazione altrettanto semplice di quella degli spettri di assorbimento sulla base della legge di Lambert-Beer . Il metodo è quindi chiamato spettroscopia di rifrazione infrarossa. Per gli spettri di riflessione di materiali inorganici con forti oscillatori, viene utilizzata l'analisi di dispersione o la trasformazione di Kramers-Kronig (KKT) per calcolare lo spettro di assorbimento del campione.
Oltre al metodo sopra descritto, è adatto e adsorbito un ulteriore metodo di riflessione, specialmente per strati sottili esistenti , la cosiddetta tecnica di " spettroscopia di assorbimento a riflessione infrarossa " (Inglese: spettroscopia di assorbimento a riflessione infrarossa , IRRAS, anche IRAS). Qui la radiazione infrarossa viene riflessa su una superficie metallica (esterna) sulla quale si trova lo strato da esaminare. A causa dell'elevata riflettività dello strato metallico (> 90%), la radiazione infrarossa viene guidata due volte attraverso il campione. Inoltre, analogamente alla spettroscopia di risonanza plasmonica di superficie , il campo viene rafforzato sulla superficie del metallo, il che rende il metodo sensibile anche per i monostrati , cioè strati sottili con uno spessore dello strato di pochi angstrom ; la rilevazione di monostrati è possibile anche con altre tecniche di misura della spettroscopia infrarossa. La misurazione viene solitamente effettuata ad un'incidenza quasi radente, cioè ad un angolo di incidenza di circa 80° dalla perpendicolare. Inoltre, la sensibilità del metodo può essere aumentata mediante l'uso di radiazioni p-polarizzate . Anche in questo caso gli spettri possono essere valutati quantitativamente utilizzando il metodo dell'analisi della dispersione.
Riflessione diffusa

Con la spettroscopia a riflessione diffusa (DRIFTS) è possibile esaminare solidi grezzi e polveri senza pressare questi ultimi in una matrice (vedi misure di trasmissione). Il raggio infrarosso è diretto sul campione e, a causa della superficie ruvida, non è più diretto (come con uno specchio), ma riflesso diffusamente . La radiazione riflessa viene nuovamente impacchettata da una speciale disposizione a specchio, emisferica sopra il campione (vedi specchio concavo ) e guidata al rivelatore.
A causa della riflessione diffusa, le componenti della radiazione sono distribuite in modo non uniforme e lo spettro risultante ha peculiarità che consentono un confronto diretto con gli spettri di altre tecnologie IR solo in misura molto limitata. Per una valutazione (qualitativa e quantitativa), lo spettro può essere convertito in uno spettro di assorbimento utilizzando la funzione Kubelka-Munk .
Riflessione interna totale attenuata (ATR)

Nella spettroscopia infrarossa ATR, la radiazione è guidata in riflessione totale in un elemento di riflessione (quasi una guida d'onda ottica ) . All'interfaccia dell'elemento si forma un campo evanescente, la cui ampiezza decade esponenzialmente perpendicolarmente all'interfaccia ( onda evanescente ). Se c'è un campione dietro l'interfaccia, il campo evanescente interagisce con il campione, cioè alcuni intervalli di frequenza possono essere assorbiti dal campione. Le aree assorbite sono ora assenti nello spettro del raggio riflesso che viene guidato al rivelatore. Poiché il campo decade molto rapidamente, i campioni per la misurazione devono essere portati molto vicino alla superficie per ottenere intensità di banda che possono ancora essere valutate. L'area misurata di solito copre solo i primi micrometri del campione. Il risultato sono spettri simili a quelli della spettroscopia di trasmissione, che, tuttavia, presentano differenze di intensità dipendenti dalla lunghezza d'onda in confronto. Questo metodo è adatto per campioni solidi e liquidi con assorbimenti relativamente deboli, che sono accompagnati solo da piccole variazioni dell'indice di rifrazione, altrimenti il campo elettrico non è più evanescente nel campione.
emissione

Nella spettroscopia di emissione infrarossa (engl. Spettroscopia di emissione infrarossa , IRES) di un campione viene emessa la radiazione infrarossa studiata. Ciò significa che, a differenza dei metodi di spettroscopia di assorbimento sopra descritti, il campione non viene irradiato da una sorgente luminosa ad alta temperatura e confrontato con uno spettro di riferimento senza campione, ma il campione stesso funge da sorgente di radiazione. Gli spettri di emissione ottenuti mostrano significativamente meno dettagli per quanto riguarda gli spettri rotazionali e l'intensità e la forma della banda dipendono maggiormente dalla temperatura.
La misura di campioni privi di spettro di riferimento è possibile perché una molecola che si trova in uno stato di oscillazione eccitata può emettere radiazione infrarossa con una certa probabilità . Le bande di emissione misurate corrispondono solitamente alla transizione da uno stato eccitato a uno stato fondamentale. L'emissione può avvenire sia in presenza (per emissione indotta ) sia in assenza (per emissione spontanea ) di radiazione elettromagnetica incidente. Tuttavia, a temperatura ambiente il numero di molecole che si trovano in uno stato eccitato è relativamente piccolo (meno dell'1% del numero allo stato fondamentale, basato su un valore limite a 1000 cm −1 ) e quindi significativamente inferiore rispetto al caso di assorbimento. Per ottenere un segnale più forte si riscalda solitamente un campione (solitamente sopra i 100°C) e si misura la radiazione emessa (vedi corpo nero ). Inoltre, sulla base del prodotto del fattore di Boltzmann e della probabilità di transizione, ci si possono aspettare solo energie di radiazione molto basse, per cui la spettroscopia IRE è stata molto complessa per lungo tempo e ha mostrato solo una bassa sensibilità. Con interferometri migliorati e nuovi rivelatori sensibili, gli spettri di emissione possono ora essere registrati regolarmente.
La spettroscopia IRE viene utilizzata, tra l'altro, per esaminare vernici , sali fusi e silicati , nonché reazioni catalitiche su strati sottili e specie adsorbite su solidi. Questo metodo offre vantaggi soprattutto per le misure in situ ad alte temperature, poiché qui, a differenza, ad esempio, delle tecniche a riflessione, la sovrapposizione delle bande di assorbimento con le bande di emissione è trascurabile. La spettroscopia IRE può essere utilizzata anche per misurare grandi volumi di gas, ad esempio nell'analisi di processo .
Le misurazioni di più strati o di volumi generalmente più grandi possono essere problematiche, poiché qui si verificano processi di assorbimento e di emissione-riassorbimento che influenzano gli spettri misurati. Altri parametri che influenzano lo spettro sono la geometria del campione, la scelta del fondo di radiazione, la radiazione diffusa e, come già detto, la temperatura del campione stesso.Per questi motivi anche spettri di emissione IR con un elevato rapporto segnale-rumore rapporto sono molto complessi e difficili da valutare. Pertanto, in letteratura sono stati presentati vari metodi per compensare l'interferenza.
Guarda anche
- Chimica di superficie
- Divieto alternativo
- globale
- Principio di Franck-Condon
- Journal of Near Infrared Spectroscopy
letteratura
- Helmut Günzler, Hans-Ulrich Gremlich: Spettroscopia IR: un'introduzione . 4a edizione. Wiley-VCH, Weinheim 2003, ISBN 978-3-527-30801-9 .
- M. Hesse, H. Meier, B. Zeeh: metodi spettroscopici in chimica organica . 6a edizione. Thieme, Stoccarda 2002, ISBN 3-13-576107-X .
- Bernhard Schrader : spettroscopia infrarossa e Raman . VCH, 1995, ISBN 3-527-26446-9 .
link internet
- Nozioni di base e tecnologia della spettroscopia a infrarossi
- Database spettrale per composti organici, SDBS (inglese)
- Raccolta di vari spettri IR
- Spettroscopia IR e chimica organica (inglese)
- Spettri di vibrazione rotazionale a Chemgapedia
- Elenco delle banche dati e delle opere di riferimento dell'ETH di Zurigo con spettri IR
- Elenco delle banche dati e delle opere di riferimento dell'ETH di Zurigo con spettri IR lontani
- Elenco delle banche dati e delle opere di riferimento dell'ETH di Zurigo con spettri nel vicino IR
Evidenze individuali
- ↑ Spettroscopia infrarossa BGR. Estratto il 17 luglio 2008 .
- ↑ a b Alexander Erlich: Spettroscopia infrarossa in trasformata di Fourier. ( Memento dell'11 dicembre 2014 in Internet Archive ) (Esempi per spettri IRES; PDF; 2.6 MB), 8 luglio 2010.
- ↑ Helmut Günzler, Hans-Ulrich Gremlich: Spettroscopia IR: un'introduzione . 4a edizione. Wiley-VCH, Weinheim 2003, ISBN 978-3-527-30801-9 , pp. 165-240 .
- ^ Thomas Günter Mayerhöfer, Susanne Pahlow, Jürgen Popp: La legge Bouguer-Beer-Lambert: Brillare l'oscurità . In: ChemPhysChem . n/a, n/a, 14 luglio 2020, ISSN 1439-4235 , doi : 10.1002 / cphc.202000464 .
- ^ Thomas G. Mayerhöfer, Susanne Pahlow, Uwe Hübner, Jürgen Popp: Rimozione degli effetti basati sull'interferenza dagli spettri infrarossi: rivisitazione delle frange di interferenza . In: Analista . nastro 145 , n. 9 , 4 maggio 2020, ISSN 1364-5528 , pag. 3385-3394 , doi : 10.1039 / D0AN00062K ( rsc.org [consultato il 10 gennaio 2021]).
- ↑ C. Christiansen: Indagini sulle proprietà ottiche di corpi finemente suddivisi - prima comunicazione . In: Annali di Fisica e Chimica . 23, 1884, pp. 298-306.
- ↑ C. Christiansen: Indagini sulle proprietà ottiche dei corpi finemente suddivisi - seconda comunicazione . In: Annali di Fisica e Chimica . 24, 1885, pp. 439-446.
- ↑ CV Raman : La teoria dell'esperimento di Christiansen . In: Proc. Ind. Acad. Sci. 29, 1949, pp. 381-390.
- ↑ a b c d (PDF) Ottica d'onda nella spettroscopia infrarossa. Consultato il 21 agosto 2021 .
- ^ Thomas G. Mayerhöfer, Vladimir Ivanovski, Jürgen Popp: Spettroscopia di rifrazione infrarossa . In: Spettroscopia applicata . 18 agosto 2021, ISSN 0003-7028 , pag. 000370282110367 , doi : 10.1177 / 00037028211036761 ( sagepub.com [consultato il 21 agosto 2021]).
- ^ Thomas Mayerhöfer: Ottica dell'onda nella spettroscopia infrarossa . 2021, doi : 10.13140 / RG.2.2.14293.55520 ( rgdoi.net [consultato il 26 aprile 2021]).
- ↑ a b W. Suetaka: Infrarosso di superficie e spettroscopia Raman: metodi e applicazioni . Springer, New York 1995, ISBN 0-306-44963-3 , pp. 163 ff .
- ^ S. Zhang, FS Franke, TM Niemczyk: Spettroscopia di emissione . In: Francis Mirabella, Mirabella (Ed.): Tecniche moderne in spettroscopia molecolare applicata . Wiley & Sons, 1998, ISBN 0-471-12359-5 , pp. 323-376 (introduzione con descrizioni di varie tecniche).
- ↑ K. Molt: Registrazione di spettri di emissione infrarossa di strati sottili su superfici metalliche con l'aiuto di uno spettrometro a reticolo accoppiato a computer . In: Journal for Analytical Chemistry di Fresenius . nastro 308 , n. 4 , 1981, pp. 321-326 , doi : 10.1007 / BF00516246 (con riferimento a G. Herzberg: Spectra of biatomic molecole. Van Nostrand, Princeton 1950.).
- ^ Francis M. Mirabella: Tecniche moderne nella spettroscopia molecolare applicata . Wiley-IEEE, 1998, ISBN 978-0-471-12359-0 , 9° spettroscopia di emissione, pp. 323-377 .